过去10年间,ICH成员组织(美国FDA、欧盟EMA和日本PMDA)的监管环境都有很大进步,新药审批时间有所减少,更多的新药进入市场为患者所用。此外,多元化监管途径和方式的引入、规范化和广泛应用在此进程中扮演了重要的角色,尤其对开发急需的药品的审批发挥了重要的作用。本文分析了ICH的三个主要成员组织2005~2014年间监管的趋势变化。
2014年是过去10年中批准新分子(NASs)最多的一年,PMDA是在3个监管机构中批准NASs最多的一个(表1)。
表1.2005~2014年间FDA、EMA、PDMA批准的NASs数量
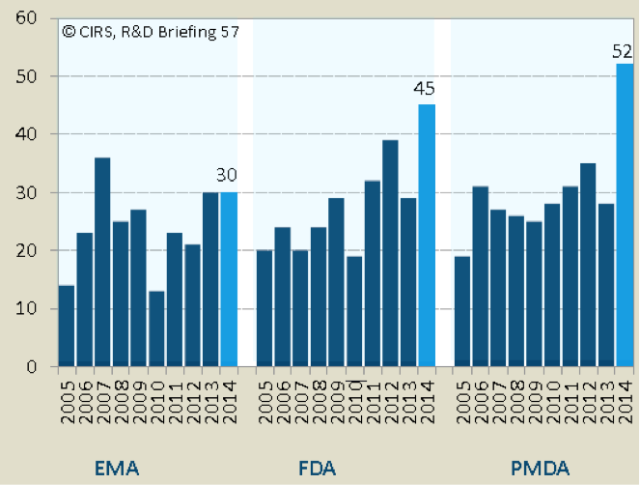
2013年新批准的NASs数量有所减少,在此基础上,FDA和PMDA2014年批准的NASs数量同比分别增长了55%和86%。虽然PMDA批准的数量最多,但其中63%的混合物之前已被FDA和EMA批准(表5)。
2005~2009和2010~2014年之间,FDA和PMDA新批准的NASs数量分别增长了40%、36%,EMA的批准数量也在增长,但低于FDA的增长率。许多FDA在2014年批准的NASs正在通过EMA的审批或者已在2015年批准。
2005~2014年间FDA和PMDA批准NASs的平均时间,2014年PMDA是速度时间最短的(表2)。
表2. 2005~2014年间FDA和PMDA批准NASs的平均时间
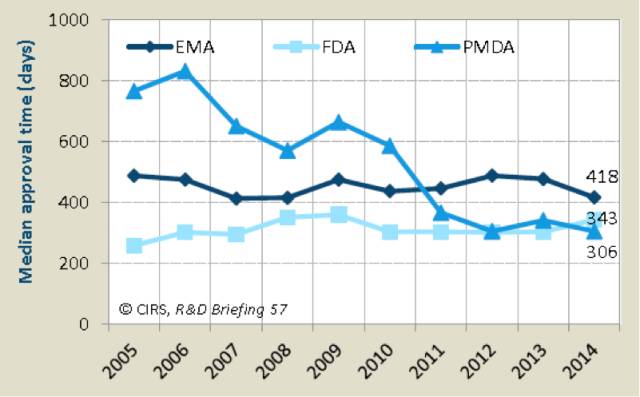
(注:EMA批准时间包括欧盟委员会时间)
历史上日本新药的批准时间相对较长,但随着PMDA资源的增加和更多的承诺,PMDA的审批时间目前已与FDA相当。
2014年,FDA的平均审批时间5年来首次加快(加速大约40天),这可能是引入仿制药申请者付费法案(PDUFA)带来的程序变化。在现有的法规和程序框架下,2011年起欧洲便是在三个地区中审批时间最长的一个,但2014年也是EMA过去10年中审批时间最短的一年。
2014年,快速审评(即EMA的审评加速程序和FDA/PMDA的优先审评程序)分别占到了FDA、PMDA和EMA58%、50%和13%的份额(表3)。
表3.2005~2014年检ICH成员组织审评类型统计
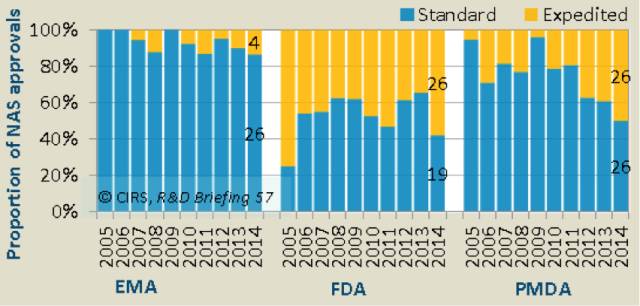
过去十年中,FDA和PMDA的审评体系日益完善,快速审评手段的推出促进了创新药的诞生。过去十年中,FDA通过的快速审评大约占到总体数量的47%左右,而PMDA在此期间的快速审评增加了一倍。PMDA作出变化主要是为了满足日本患者对新药的需求。
EMA使用加速审评渠道较少,这说明EMA对加速审评所要求的标准比FDA和PMDA严格,程序更加复杂,2014年,有3个NASs申请是通过快速审评通道审批,但是在审评过程中又回到了普通审评通道。尽管快速审评通道的使用方式不同,但2014年3个监管机构的平均审批时间差别不大,也就是说EMA的快速审评迎合了减少审批时间的趋势,但是不经常使用。
尽管快速审评的利用存在差异,ICH成员组织的结果显示2014年这些审评的平均通过时间类似(表4)。
表4. 2005~2014年检ICH成员组织通过不同审批渠道审批NASs平均时间统计
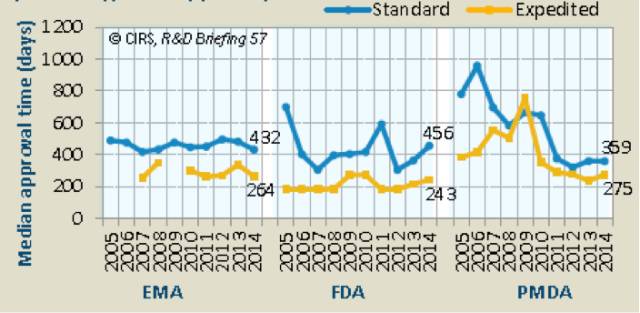
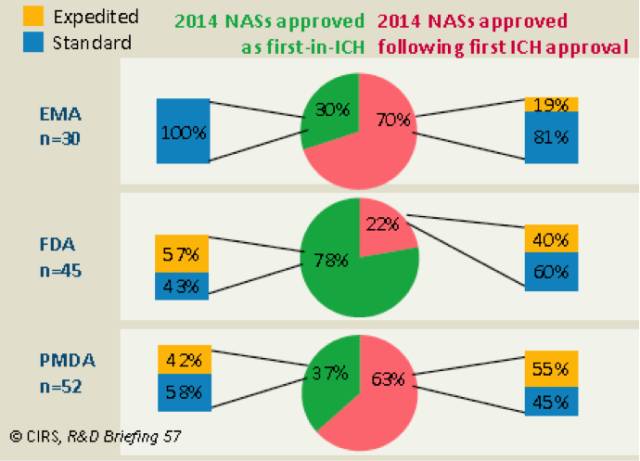
三个监管机构的审批平均时间不同之处在于,2012年起,FDA的标准审评时间增长了150天,快速审评增长了60天,部分原因是PDUFA V给整个审评过程增加了两个月的时间。在2014年三个机构批准的所有NASs中,FDA是首次批准数量最多的组织,占比78%,EMA和PMDA分别占比30%和37%。
对于2014年被各机构通过的NASs,相比于EMA和PMDA,FDA被first-in-ICH品准的比例最高。
表5. 2014年ICH成员组织不同类型审批所需时间
孤儿药审批
2014年,ICH成员组织孤儿药的审批数量都创下了十年来的新高(表6)。
表6. 2005~2014年ICH成员组织批准的孤儿药所占比例
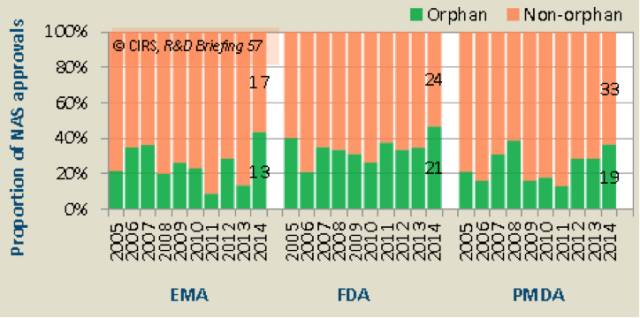
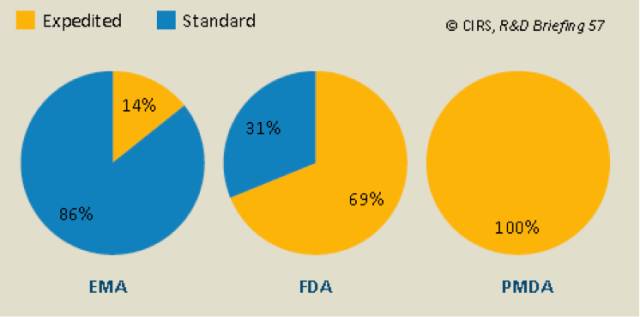
2014年EMA、FDA、PMDA所批准的孤儿药分别占43%、47%和37%,与2005~2009,2010~2015年相比,孤儿药所占比重变化不大,EMA下降了5%,FDA和PMDA分别增长了5%和2%。2014年孤儿药审批的增多是一次性事件还是长期的趋势,需要更多的时间去观察。2010~2014年,快速审查用于孤儿药在EMA、FDA、PMDA所占的比重分别为14%,69%和100%。
2010-2014年,EMA,FDA和PMDA的孤儿药分别使用了14%、69%和100%的快速审评(表7)。
表7. 2010~2014年ICH成员组织孤儿药的审查类型
除了快速审查,其他加速审评的渠道(EMA特别环境和条件下审批,FDA的快速通道、优先审评和突破性疗法资质认定)使得孤儿药能够更加快速与患者见面。
看一下2010~2014年孤儿药使用快速通道审查的比率会发现,43%通过EMA审批的孤儿药是通过三种快速通道之中的一种审批,非孤儿药为17%;85%通过FDA审批的孤儿药通过FDA四种快速通道之一的一种审批,非孤儿药为39%;而通过PMDA审批的全部孤儿药都是通过快速审查通道。
过去10年,FDA和PMDA审批通过孤儿药的平均时间都要快于非孤儿药,EMA的审批时间则大致相似(表8)。
表8. 2005~2014年ICH成员组织审评孤儿药所需平均时间
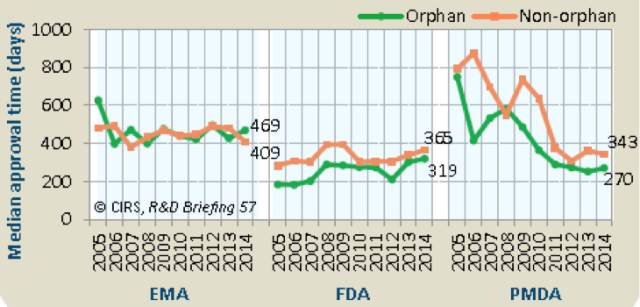
2014年FDA和PMDA审批非孤儿药的时间要比审批孤儿药的时间短46和73天,这主要是由于孤儿药审批使用快速通道所致。EMA孤儿药的审批时间要少60天。
表9. ICH成员组织年度审批通过的各治疗领域NASs
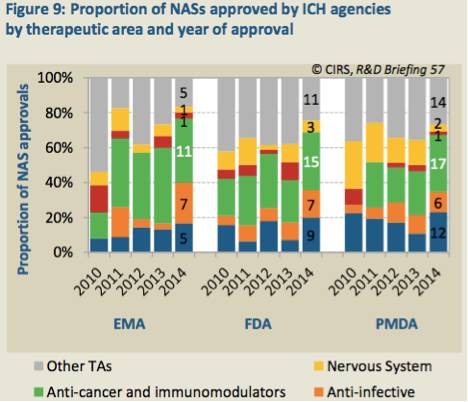
表10. ICH成员组织2010~2014年审批通过的各治疗领域NASs及所使用的审评通道
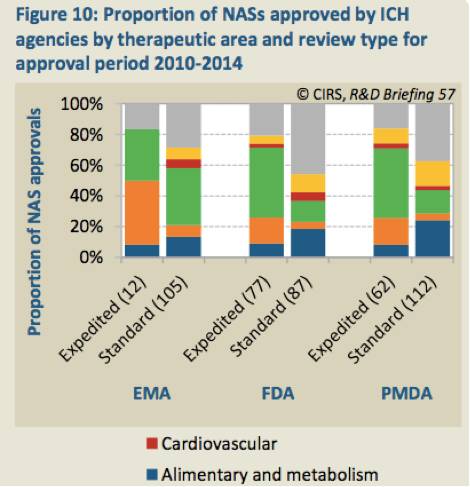
(注:从图中可以看出,2010~2014年,抗癌和免疫调节类NASs通过FDA和PDMA审评所占比重最大,EMA是抗感染类NASs。)
消化系统、抗感染、免疫系统等治疗领域的NASs2014年批准的最多。从审查通道来看看,FDA和PMDA在各治疗领域标准通道和快速通道的审查数量大体一致。EMA有所不同,在过去的5年中,在审批的抗癌药中每43个中有4个通过快速审查通道,而FDA和PMDA的比例分别为35/47和28/45。
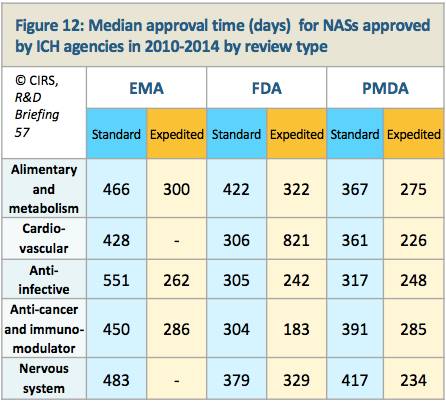
就FDA和PMDA而言,比较不同治疗领域审批时间的中位值,可以看到,抗感染、抗肿瘤和免疫调节剂药物获得加速审评的比例较高(表11和表12)。
表11. 2010~2014年不同类型治疗领域NASs的批准时间
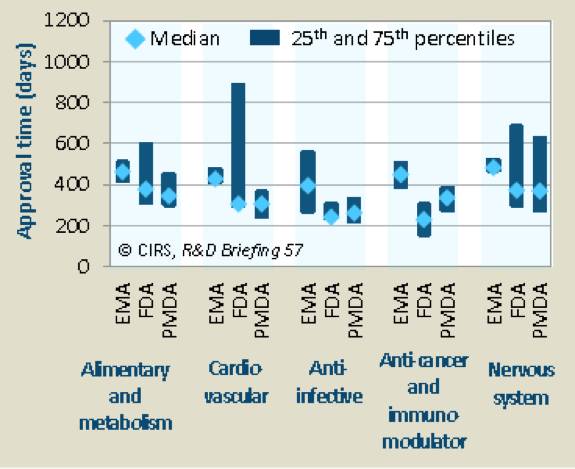
表12. 2010~2014年ICH成员组织各治疗领域NASs审批平均时间(按照审查类型)
2010年至2014年期间,在3家ICH机构共批准了35个NAS。但是不同机构提交滞后时间、批准类型和速度各不相同(表13和表14)。
表13:2010年至2014年期间所有ICH成员国批准的35个新活性物质(NAS)的提交滞后时间中位值

表14:2010年至2014年期间所有ICH成员国批准的35个新活性物质(NAS)不同审批类型的批准时间中位值
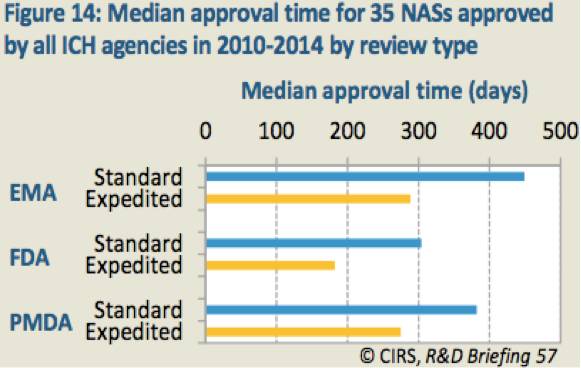
EMA和FDA的申请提交基本同步,但是PMDA要晚将近1年。FDA的批准速度最快,接下来依次是PMDA和EMA。FDA、PMDA和EMA的加速审评比例分别为57%、54%和11%。
在获批的35个普通批准中,9个是在2013-2014年批准的(最后1个批准是在2014年)。获得FDA、PMDA和EMA加速审评的产品分别是5个、4个和1个,不过这几个产品在审评过程中被退回成标准审评。但这再一次展示了在EMA审批体系里获得加速路径的标准或流程限制(表15)。
表15:2013-2014所有ICH机构批准的10个NAS的批准情况,最后获批发生在2014年
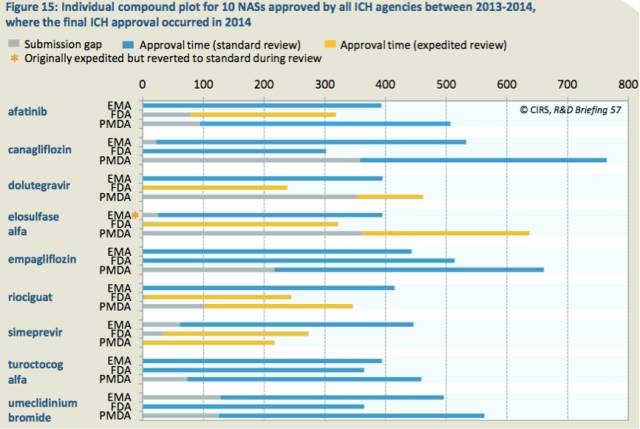
2013-2014年批准的9个产品中,7个基本上同时向EMA和FDA提交申请(30天之内)。除了1个药物(simprevir)首先在日本提交申请(比FDA提前34天)外,其他8个都是最后向PMDA提交申请的,时间差中位值171天。这说明,虽然自2010-2011年起,日本的提交滞后时间已经在缩短,但仍旧有延迟(表22)。
2014年,EMA总体批准时间中位值减少,主要原因是公司回应时间的减少(表16)。
表16:每年EMA NAS批准审评流程的中位值
表17:2010至2014年期间EMA不同审评类型NAS批准审评流程的中位值
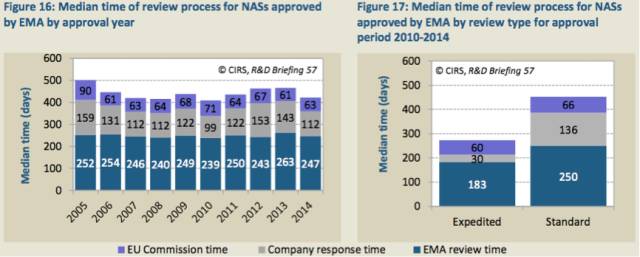
与2013年相比,2014年公司回应时间的中位值减少了31天;同期EMA的审评时间中位值减少了11天,欧盟委员会所花费的时间不变。比较2010至2014年期间加速审评和标准审评的批准时间(表17),发现加速审评时公司的回应时间快了4.5倍。这是由于公司的回应截止时间被规定了,如果超出一个月,EMA可以作出决定恢复成标准审评。EMA加速审评的时间比标准审评快1.4倍,原因是CHMP的审评时间更短(150天,而不是210天)。然而,无论哪种审评类型,欧盟委员会的审批时间相同,这也为欧盟委员会未来加快产品审批提供了可能。
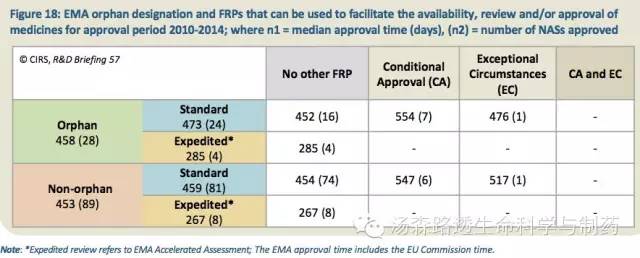
2010至2014年期间,获批的NAS中35%(41/117)受益于三种加速监管路径(FRP)中的至少一种,或者EMA孤儿药指定,以加速审评和/或批准及上市(表18)。
除了加速审评(指的是EMA的加速评估),目前还有两种其它的FRP,有条件批准(CA)和特殊情况(EC)。然而,对于指定为CA的产品,批准时间比标准的EMA审评时间中位值要长大约100天,其原因已经在2014年Escher的报告里指出,这个路径是作为“挽救路径”,用于那些不能通过标准审批路径批准的产品,而不是一个“预先计划的提早上市”的路径。2010至2014年期间和自由2个NAS使用EC指定。不过,EMA当前正在通过实验项目,探索新的可行的提前批准的路径。
表18:2010至2014年期间获得EMA的孤儿药指定和FRP的药物,这两个路径可用于加速审评和/或批准及上市;n1=批准时间的中位值(天),(n2)=批准的NAS数量
2005-2009年期间到2010-2014年期间,FDA药品评价和研究中心(CDER)仅经过一个审评周期就批准的NAS数量从68%增长至76%(表19)。2010-2014年期间,与标准审评流程相比,加速流程中审评周期为1个的所占的比例更大(表20)。
表19:每年经过不同审评周期数获得CDER批准的NAS数量
表20:2010至2014年间CDER按照不同审评类型批准的NAS中不同审评周期所占比例
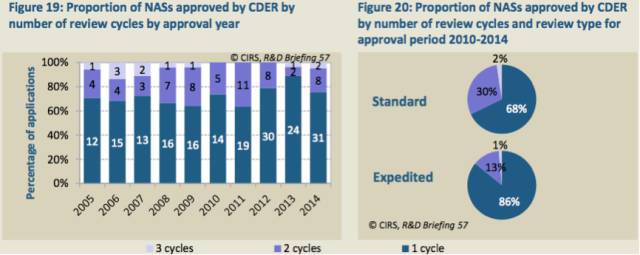
CDER正在设法进一步地优化审评流程,特是增加一个审评周期批准的产品的数量。这种在审评周期数量上的改进可能会要求更好的文件质量,反过来对审评效率起到积极的影响。但需要重视的是,这个分析(表19)仅包括已经批准的产品,若将未批准的产品统计进去,也许会出现不同的结果。
2010至2014年期间,在三个监管机构中,FDA最多地使用FRP来加速药物的审评及上市批准,以满足未被满足的医疗需求;62%(101/164)的NAS享受到4种FRP途径中的至少1种,或者获得FDA的孤儿药指定(表21)。
表21:2010至2014年期间,FDA孤儿药指定和FRP,这些指定可以加速药物的审评和/或批准及上市;n1=批准时间中位值(天),(n2)=NAS批准数量
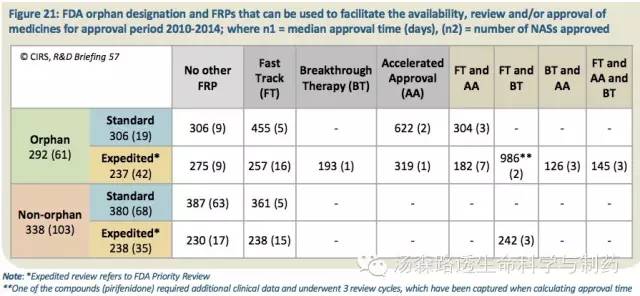
除了加速审评(参照FDA优先审评Priority Review),FDA还提供另外3种FRP,分别是:快速通道指定(Fast Track,FT)、加速批准路径(Accelerated Approval,AA)以及2012年引入的突破性疗法资格(Breakthrough Therapy,BT)。虽然FT和AA本身并不能导致更快的审评,但是可以与加速审评相结合。与仅符合加速审评标准的NAS相比,这些受益于FT和AA的申请通常审批时间会更加快。比如,孤儿药就可以获得多种FRP,通常批准更快。可能是因为这个事实,一些FRP也会促使机构与申办方之间的交流更加频繁且开始得更早(在之后的审评阶段这可能会导致被提出的问题更少),为滚动提交提供可能。
虽然与2013年相比,2014年日本药物申请的提交滞后时间增长了2.5倍,但是,2010至2014年期间,整体上,提交滞后时间在大幅下降(表22)。这看上去与公司所在国以及审评类型有关,2014年,非日本公司的孤儿药产品的提交滞后时间是最长的(表23-25)。此外,2014年日本批准的一些产品是通过政府项目加速获批的。
表22:每年日本批准的NAS的提交滞后时间*和批准时间
表23:不同国家公司和批准年份获得日本批准的NAS的提交滞后时间*和批准时间
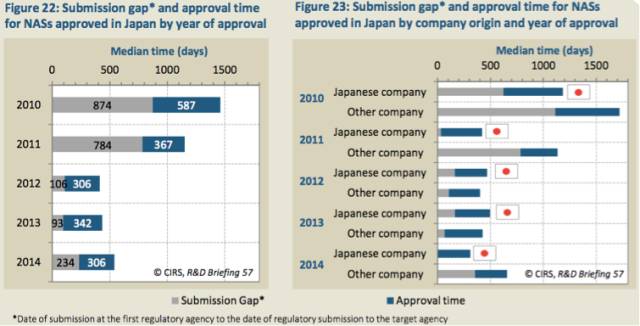
表24:2014年日本批准的NAS,不同公司所在地和不同审评类型的提交滞后时间*和批准时间
表25:2014年日本加速批准的NAS,不同公司所在地和不同审评类型的提交滞后时间*和批准时间
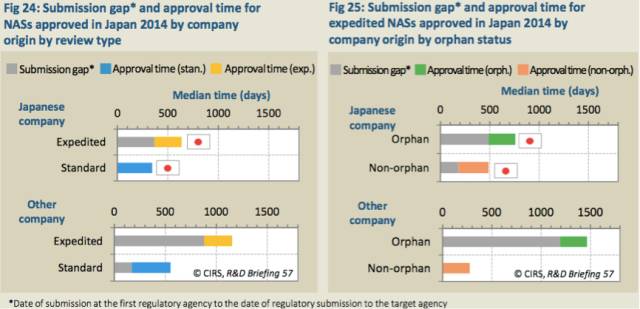
比较2005-2009年和2010-2014年这两段时间,日本公司获得批准的NAS数量占全部获批NAS数量的比例从39%增长至51%。这些公司通常是立足于为日本人群开发产品,所以与非日本的公司相比,提交滞后时间相对较短。非日本的公司一般首先去寻求EMA和FDA的批准。除了公司所在地,审评类型(和孤儿药地位)也影响着提交滞后时间。不考虑公司所在地,孤儿药比非孤儿药的提交滞后时间更长。
批准时间
申请提交至监管机构批准之间所花的时间。这个时间包括监管机构和公司所用的时间。注:EMA的批准时间包括欧盟委员会的审查时间。
生物制品
从动物组织分离出的物质,或者通过重组DNA或杂交瘤技术在细胞系、转基因动物或转基因植物表达而获得的产品,用于人类的治疗、预防或体内诊断。
化学实体
通过化学合成产生的实体物质。
加速审评(Expedited review)
简单地说,加速审评指的是EMA的加速评价(Accelerated Assessment)以及FDA和PMDA的优先审评(Priority Review)。
加速法规路径(Facilitated Regulatory Pathway,FRP)
用以加速审评和/或批准及上市药品的监管路径,为未被满足医疗需求的药品提供一条标准审评路径之外的替代途径。
新活性物质(New active substances,NAS)
化学、生物、生物技术或放射性物质,之前没有用作人类治疗,将作为“处方药”提供,可用于人类疾病的治愈、缓解、治疗、预防或体内诊断。
NAS还包括:
之前已上市化学药物盐类的一种异构体、异构体的混合物、复合物或衍生物,与之前的药物在安全性和有效性上不同。
之前已上市作为药品的生物制品,但是分子结构、来源或者生产工艺不同,需要进行临床研究。
放射性物质,之前没有用作药品的放射性核素或配体。或者,分子和放射性核素连接的机制之前没有使用过。
以下申请被排除在外:
疫苗
提交新的临床数据的其它申请。
仿制药申请。
已经获批并用于同一适应症的产品,另一家公司提交的完全新的文档。
已经存在的化合物,以一个新的或额外的名称,或改个名字申请(即“克隆”申请)。
提交滞后时间(Submission gap)
申请提交至首个监管机构与提交至目标监管机构之间的时间差。
WHO ATC分类
A - 消化道和代谢:用于酸紊乱性疾病药物、胃肠功能紊乱药物、止吐药、治疗恶心的药物、胆和肝病药物、泻药、止泻药、肠道抗炎或抗感染药,糖尿病药物。
C - 心血管药物:心脏病药物、抗高血压药物、β阻断剂、钙通道阻滞剂、作用于肾素-血管紧张素系统的药物和降低血脂的药物。
J - 抗感染药物:全身使用的抗菌药、全身使用的抗真菌药、抗真菌细菌药、全身使用的抗病毒药、免疫血清和免疫球蛋白,疫苗。
L - 抗癌和免疫调节药:抗肿瘤药物、内分泌治疗药物、免疫增强剂、免疫抑制剂。
N - 神经系统药物:麻醉药、镇痛药、抗癫痫药、抗帕金森病药、精神阻断剂、精神兴奋剂,其他神经系统药物。
本文出自:Centre for Innovation in Regulatory Science(CIRS)CIRS-药政科学创新中心,是汤森路透旗下的一家位于英国的独立运营分支机构,隶属于汤森路透知识产权与科技部。