



Add the following:
PURPOSE
This general chapter presents a framework for the design, justification, and implementation of assessments for drug product leachables derived from pharmaceutical packaging and delivery systems. A scientifically sound leachables assessment is important to manufacturers and their various suppliers primarily as a means of establishing the suitability for use of pharmaceutical packaging/delivery systems, as leachables can potentially affect drug product efficacy, safety, and quality. Additionally, such a leachables assessment could provide an understanding of the sources of leachables and how to evaluate and manage leachables during the drug development and manufacturing processes. The chapter establishes critical dimensions of a leachables assessment and discusses practical and technical aspects of each dimension. The chapter does not establish specific analytical methods or leachables specifications and acceptance criteria for any particular dosage form or packaging system or drug product combination; nor does it delineate every situation in which a leachables assessment is required. It is not possible for a general discussion of drug product leachables to anticipate and cover all situations which can occur in the pharmaceutical industry where a leachables assessment might be required. Designing an individual leachables assessment is a process that strikes a balance between sound science, prudent resource allocation, and effective risk management with an emphasis on patient safety and product quality. Achieving this balance is the responsibility and obligation of the drug product manufacturer, and assumes due consideration of applicable legal and regulatory requirements. The principles and best demonstrated practices outlined in this general chapter represent a consensus interpretation of sound science and can therefore be extrapolated and applied to any situation in which a leachables assessment is required for pharmaceutical application.
In many cases, drug product leachables assessments are based on or facilitated by knowledge from extractables assessments accomplished on drug product packaging systems, packaging components, and packaging materials of construction (see Assessment of Extractables Associated with Pharmaceutical Packaging/Delivery Systems  1663
1663 ).
).
KEY TERMS
This general chapter uses the following key terms (1,2; also see Packaging and Storage Requirements 659). Note that the terms Packaging System, Packaging Component, Primary Packaging Component, Secondary Packaging Component, and Materials of Construction are also defined in 659, and the definitions below are intended for clarification purposes within the context of this chapter and are not intended to supersede those provided in 659.
Packaging Systems are the sum of packaging components that together contain and protect the dosage form. Packaging systems are also referred to as Container Closure Systems and may include primary, secondary, and tertiary packaging.
A Container is a receptacle that holds an intermediate compound, active pharmaceutical ingredient, excipient, or dosage form and is in direct contact with the product.
A Closure is a material that seals an otherwise open space on a container and provides protection for the contents. It also provides access to the contents of the container.
A Packaging Component is any single part of the package or container–closure system including the container (e.g., ampuls, prefilled syringes, vials, bottles), closures (e.g., screw caps, stoppers), ferrules and overseals, closure liners, inner seals, administration ports, overwraps, adminstration accessories, labels, cardboard boxes, and shrink wrap.
A Primary Packaging Component is in direct contact or may come into direct contact with the product (e.g., IV bag).
A Secondary Packaging Component is in direct contact with a primary packaging component and may provide additional protection of the product (e.g., overpouch or dustcover for an IV bag).
A Tertiary Packaging Component is in direct contact with a secondary packaging component and may provide additional protection of the product during transportation and/or storage (e.g., shipping carton for an overpouched IV bag).
An Ancillary Component is a component or entity that may come into contact with a tertiary packaging component during the distribution, storage, and transportation of the packaged product (e.g., pallets, skids, shrink wrap, active containers).
Packaging Materials of Construction are substances used to manufacture packaging components. These are also referred to as Raw Materials.
A Delivery System is the sum of components and materials that are used to transport a drug product from its packaging to the point of administration into the patient. For example, an administration set is a delivery system that is used to transfer liquid drug products from their plastic packaging system to the site of administration to the patient.
Extractables are organic and inorganic chemical entities that can be released from a pharmaceutical packaging/delivery system, packaging component, or packaging material of construction and into an extraction solvent under laboratory conditions. Depending on the specific purpose of the extraction study (discussed below), these laboratory conditions (e.g., solvent, temperature, stoichiometry, etc.) may accelerate or exaggerate the normal conditions of storage and use for a packaged dosage form. Extractables themselves, or substances derived from extractables, have the potential to leach into a drug product under normal conditions of storage and use and become leachables. Thus extractables are potential leachables.
Leachables are foreign organic and inorganic chemical entities that are present in a packaged drug product because they have leached into the packaged drug product from a packaging/delivery system, packaging component, or packaging material of construction under normal conditions of storage and use or during accelerated drug product stability studies. Because leachables are derived from the packaging or delivery system, they are not related to either the drug product itself or its vehicle and ingredients. Leachables are present in a packaged drug product because of the direct action of the drug product on the source of the leachable. Thus leachables are typically derived from primary and secondary packaging, because the primary and secondary packaging can serve as a barrier between the packaged drug product and other potential sources of foreign chemical entities (e.g., tertiary packaging and ancillary components). In certain circumstances, packaging may directly contact the patient under typical clinical conditions of use (e.g., the mouthpiece of a metered dose inhaler). As a result of this contact, patients may be exposed to leachables from the packaging without the action of the drug product. Leachables are typically a subset of extractables or are derived from extractables. Note that chemical entities can also migrate from packaging/delivery systems to patients via direct contact.
Migrants are also foreign organic and inorganic chemical entities that are present in a packaged drug product because they have leached into the packaged drug product from a packaging/delivery system, packaging component, or packaging material of construction under normal conditions of storage and use or during accelerated drug product stability studies. However, migrants are differentiated from leachables by the circumstance that migrants accumulate in the packaged drug product after the migrant has crossed a physical barrier, such as that provided by primary and secondary packaging. Because migrants cross a physical barrier, they are not present in the packaged drug product due to direct action of the drug product on the source of the migrant because the barrier prevents such direct action. Thus migrants are derived from secondary and tertiary packaging and ancillary components.
Regardless of whether a substance is a leachable or migrant, it is still a foreign substance in the packaged drug product, and thus its impact must be assessed in the same manner. However, as the means by which a leachable and a migrant become entrained in a packaged drug product may be different, extractables studies meant to address leachables may be designed and implemented differently than extractables studies meant to address migrants.
Leachables Studies are laboratory investigations into the qualitative and quantitative nature of a particular leachables profile(s) over the proposed shelf-life of a particular drug product.
Characterization is the discovery, identification, and quantitation of each individual organic and inorganic leachable present in a drug product formulation above a predetermined level or threshold. Such thresholds should be based mainly on patient safety considerations, with consideration also given to the capabilities of analytical technology, and other related issues.
Identification is the process of assigning a molecular structure to an organic leachable, or assigning constituent elements in the case of an inorganic leachable.
Quantitation is the process of measuring the level, or concentration, of an individual organic or inorganic leachable contained in a drug product formulation.
Leachables Profiles are qualitative and/or quantitative analytical representations of the leachables content of a particular drug product formulation.
Leachables–Extractables Correlations are established when observed drug product leachables are linked both qualitatively and quantitatively to extractables from associated packaging/delivery systems, packaging components, or materials of construction.
Threshold of Toxicological Concern (TTC) is a level of exposure for all chemicals, whether or not there is specific toxicity data, below which there would be no appreciable risk to human health (6). The TTC approach is a form of risk characterization in which uncertainties arising from the use of data on other compounds are balanced against the low level of exposure.
Safety Concern Threshold (SCT) is the threshold below which a leachable would have a dose so low as to present negligible safety concerns from carcinogenic and noncarcinogenic toxic effects.
Qualification Threshold (QT) is the threshold below which a given noncarcinogenic leachable is not considered for safety qualification (toxicological assessments) unless the leachable presents structure-activity-relationship (SAR) concerns.
Analytical Evaluation Threshold (AET) is the threshold at or above which a leachable should be characterized and reported for toxicological assessment. The AET can be mathematically derived from the SCT (or other threshold concepts) based on factors that include the dosing parameters of the drug product.
BACKGROUND
Management of leachables is important to pharmaceutical and biotechnology/biologic product manufacturers and regulatory authorities because certain leachables above specific concentrations can present safety concerns for patients and/or compatibility issues for drug product formulations. During the 1980s, the U.S. Food and Drug Administration (FDA) began to formally and comprehensively address leachables in drug products after findings of patient sensitivity induced by leachables and other potential safety concerns related to leachables (2–4). Since then, management of both extractables and leachables for packaging systems and final drug products has become an important part of pharmaceutical development and regulatory submissions for many dosage form types, particularly for those deemed of relatively high risk for dosage form interaction with the packaging system, along with a relatively high safety risk relative to the route of administration (see Table 1). Note that Table 1 is a version of the original concept that appears in the FDA guidance Container Closure Systems for Packaging Human Drugs and Biologics (1), in which certain dosage forms in the above guidance have been downgraded to having lower potential for interaction with packaging components. Remaining relatively high-risk dosage forms include: inhalation aerosols and solutions, injectables and injectable suspensions, ophthalmics, and transdermal ointments and patches. It is important to note, however, that even low-risk dosage forms present some risk and that appropriately rigorous leachables assessments can be important to particular drug products in lower risk dosage form categories (e.g., topicals and oral dosage forms, etc.).
| Examples of Packaging Concerns for Common Classes of Drug Products |
|||
|---|---|---|---|
| Degree of Concern Associated with the Route of Administration |
Likelihood of Packaging Component-Dosage Form Interaction |
||
| High |
Medium |
Low |
|
| Highest |
Inhalation Aerosols and Sprays |
Injections and Injectable Suspensions; Inhalation Solutions |
Sterile Powders and Powders for Injection; Inhalation Powders |
| High |
Transdermal Ointments and Patches |
Ophthalmic Solutions and Suspensions; Nasal Aerosols and Sprays |
— |
| Low |
Topical Solutions and Suspensions; Topical and Lingual Aerosols; Oral Solutions and Suspensions |
— |
Oral Tablets and Oral (Hard and Soft Gelatin) Capsules; Topical Powders; Oral Powders |
|
a
While this table provides a convenient overview of the general level of regulatory concern with various dosage forms regarding leachables, it should not be inferred that “low-risk” dosage forms (e.g., oral tablets) by that definition carry no risk for leachables issues.
|
|||
This chapter will describe scientific principles and best practices for the assessment of drug product leachables, and will cover various important concepts, including: 1) the requirement for leachables studies; 2) fundamental concepts for leachables studies; 3) the basis of thresholds for leachables and general guidance about application of these thresholds; 4) design and implementation of leachables studies; 5) leachables method development and validation; 6) correlation of results from extractables assessments and routine extractables testing with leachables studies; and 7) establishment of leachables specifications including acceptance criteria.
These scientific principles and best practices apply to all organizations and individuals involved in the manufacture, marketing, and qualification of drug products and in their stability studies, including but not limited to:
-
Manufacturers of drug products for human and veterinary use where manufacturing may involve operations at the applicant holder's facilities (i.e., facilities that belong to the holder of an approved New Drug Application or Abbreviated New Drug Application) or at those of a contractor for the applicant holder
-
Manufacturers of combination drug products
-
Packaging operations by the manufacturer or a designated contractor for the applicant holder
-
Repackaging operations in which the drug product may be owned by an organization other than the primary manufacturer.
Although it is ultimately the drug product applicant's responsibility to ensure that appropriate leachables assessments are completed, manufacturers and fabricators of pharmaceutical packaging/delivery systems, packaging components, and materials of construction should also apply these scientific principles and best practices as appropriate, and applicants are encouraged to work with component manufacturers and fabricators to this end.
CONCEPTS
General Concepts for Leachables Assessment
During the course of manufacturing, packaging, storage, distribution, and administration, dosage forms and/or their formulation constituents contact components and materials of construction of manufacturing and packaging equipment, and primary and secondary packaging components and systems. Such contact may result in interactions between the dosage form and these components and materials. One such interaction is the migration, or leaching, of substances from any of these components and materials into the dosage form. Leachables, which can include both organic and inorganic (i.e., elemental) chemical entities with wide chemical diversity, are of concern due to their potential safety risk to patients and potential compatibility risks for the drug product. In order to assess these risks and manage the potential issues posed by leachables, it is necessary to know their identities and the levels to which they will accumulate in the finished drug product over its shelf-life. These two pieces of information can be used to establish the magnitude of patient exposure (dose) and therefore the safety risk posed by an individual leachable, as well as the likelihood of any drug product compatibility issues.
Regulatory guidelines, requirements, and various best practice recommendations all state that the definitive assessment of the potential impact of contact between a packaging/delivery system and a final dosage form involves testing the final drug product for leachables. In its most essential form, this impact assessment involves performing a migration, or leachables, study whose purpose is to discover, identify, and quantitate leachables that have migrated from the contacted system, components, or materials and accumulated in the finished dosage form under the product's actual manufacturing, storage and clinical use conditions. A leachables study is a laboratory investigation into the qualitative and quantitative nature of a particular leachables profile(s) over the proposed shelf-life of a particular drug product. The purpose of a leachables study is to systematically and rationally identify and quantify (i.e., characterize) drug product leachables to the extent practicable, and within certain defined analytical threshold parameters. The results of leachables studies are used in the overall leachables assessment to understand the impact of leachables on patient safety and drug product quality and stability.
Leachables studies can be used within the context of an overall leachables assessment to:
-
Facilitate the timely development of safe and effective dosage form packaging/delivery systems, manufacturing systems, and processes by assisting in the selection of components and materials of construction
-
Facilitate the establishment of qualitative and quantitative leachables–extractables correlations in drug products, when coupled with an appropriate extractables assessment(s)
-
Establish the worst-case drug product leachables profile in a manner that facilitates the development of drug product leachables specifications and acceptance criteria (should these be required), and the safety evaluation/qualification of leachables
-
Identify trends in drug product leachables accumulation levels over the shelf-life of a particular drug product
-
Facilitate change-control processes for drug product packaging/delivery systems (as appropriate), packaging components, materials of construction, formulation constituents, etc.
-
Facilitate investigations into the origin(s) of identified leachables whose presence causes out-of-specification (OOS) results for a marketed drug product.
In these ways, leachables studies and assessments can support Quality by Design (QbD) principles for the development and manufacture of pharmaceutical packaging/delivery systems and drug products.
A complete leachables assessment includes understanding the safety impact of individual leachables, safety qualification of individual leachables, and developing an understanding of the impact of individual leachables on drug product stability (i.e., compatibility) and stability. Although safety qualification is presented in general terms, the details of attaining this goal are beyond the scope of this chapter, which is limited to general scientific principles and best practices for the conduct of leachables studies and the other stated uses of the results of leachables studies within an overall leachables assessment. The reader is directed to authoritative manuscripts on this topic (2,8,9).
Note that certain packaging and combination product medical device components are (or can be) in direct contact with a patient's mouth, nasal mucosa, or other body tissue(s) during normal use of the drug product. Such packaging components include metered dose inhaler and dry powder inhaler mouthpieces, transdermal patches, etc. Patients are potentially exposed to chemical entities by direct contact from such components. Assessment of patient exposure in such direct contact scenarios is best accomplished with appropriate extractables assessments and extraction studies, and the reader is referred to 1663.
Safety Thresholds
Although leachables represent a particular class of drug product impurity, current regulatory guidance for drug product impurities specifically considers leachables to be out of scope (5). Thresholds that have been specifically proposed for drug product leachables are based on either patient safety considerations or the current capabilities of analytical technology. Safety thresholds are particularly important in a leachables assessment because current analytical technology allows detection of trace organic and inorganic chemical entities at extremely low levels (i.e., ng/mL; ng/g). Identification and risk assessment (or qualification) of every individual chemical entity in a typical leachables profile at the limits of current analytical technology is neither necessary from a toxicological perspective nor feasible in a typical drug product. Safety thresholds allow for a science- and risk-based determination of acceptable levels of leachables and can be based on established toxicological information, as well as additional safety risk factors that consider, e.g., route of administration, daily exposure, and treatment duration. Because safety thresholds are derived from exposure data they are considered in terms of units of exposure, such as Total Daily Intake (TDI). Thus, any safety threshold must be converted into units of concentration (e.g., µg/mL) so that it can be applied as an analytical threshold in the laboratory. The analytical threshold is a guide as to which chemical entities in the leachables profile should be considered for chemical characterization (i.e., confirmed identification) and safety evaluation and qualification.
An example of a safety threshold concept that has been practically applied in pharmaceutical development is the Threshold of Toxicological Concern (TTC) approach (6). The TTC concept was adopted by the European Medicines Agency (EMA) to evaluate genotoxic impurities, using an excess cancer risk factor of 10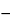 5 (1 in 100,000) (7). The EMA's proposed safety threshold for genotoxic impurities using the TTC approach is 1.5 µg/day TDI. Other examples of safety thresholds include the Product Quality Research Institute (PQRI) Safety Concern Threshold (SCT) and Qualification Threshold (QT), derived and proposed for individual organic leachables in Orally Inhaled Nasal Drug Products (OINDP) (2,8,9). The SCT is 0.15 µg/day TDI, and the QT is proposed at 5 µg/day TDI for an individual organic leachable. The development of the TTC approach provided a foundation, precedent, and guide for derivation of the PQRI SCT, which incorporates a 106 (1 in 1,000,000) risk factor rather than the 105 value used for the EMA threshold. This lower threshold was considered appropriate for leachables in OINDP because of considerations regarding the direct delivery of some of these dosage forms to diseased organs of a sensitive patient population, and assuming lifetime exposure. In addition, leachables are typically industrial chemicals with no direct structural relationship to any active ingredient or other formulation constituent. Below the SCT, identification and safety evaluation of leachables generally would not be required. Below the QT, leachables without structure alerts for carcinogenicity or irritation would not require compound-specific safety risk assessment. Note that neither the SCT nor the QT is a control threshold or safety-driven limit. Rather, they are leachables evaluation thresholds. The SCT in particular is designed to establish a threshold for characterization of unknown drug product leachables. Individual levels of safety concern, different from the SCT value, could be determined for known leachables and potential leachables (i.e., extractables).
5 (1 in 100,000) (7). The EMA's proposed safety threshold for genotoxic impurities using the TTC approach is 1.5 µg/day TDI. Other examples of safety thresholds include the Product Quality Research Institute (PQRI) Safety Concern Threshold (SCT) and Qualification Threshold (QT), derived and proposed for individual organic leachables in Orally Inhaled Nasal Drug Products (OINDP) (2,8,9). The SCT is 0.15 µg/day TDI, and the QT is proposed at 5 µg/day TDI for an individual organic leachable. The development of the TTC approach provided a foundation, precedent, and guide for derivation of the PQRI SCT, which incorporates a 106 (1 in 1,000,000) risk factor rather than the 105 value used for the EMA threshold. This lower threshold was considered appropriate for leachables in OINDP because of considerations regarding the direct delivery of some of these dosage forms to diseased organs of a sensitive patient population, and assuming lifetime exposure. In addition, leachables are typically industrial chemicals with no direct structural relationship to any active ingredient or other formulation constituent. Below the SCT, identification and safety evaluation of leachables generally would not be required. Below the QT, leachables without structure alerts for carcinogenicity or irritation would not require compound-specific safety risk assessment. Note that neither the SCT nor the QT is a control threshold or safety-driven limit. Rather, they are leachables evaluation thresholds. The SCT in particular is designed to establish a threshold for characterization of unknown drug product leachables. Individual levels of safety concern, different from the SCT value, could be determined for known leachables and potential leachables (i.e., extractables).
For OINDP there are certain “special case” compounds and compound classes, that due to particular safety concerns (e.g., carcinogenic) were deemed to require lower thresholds based on the capabilities of specific analytical technologies and methods. These special case compounds for OINDP include: polyaromatic hydrocarbons or polynuclear aromatics (PAHs or PNAs), N-nitrosamines, and the individual chemical entity 2-mercaptobenzothiozole (see Orally Inhaled and Nasal Drug Products 1664.1).
Information Sharing
To successfully manage leachables throughout the drug product lifecycle, it is critical to establish close and regular communication among those stakeholders throughout the development and drug product lifecycle responsible for the quality of the drug product: chemists, toxicologists, packaging engineers, manufacturing operations, procurement, etc. With respect to leachables, communication between the analytical chemist and toxicologist is critical. For example, if a leachable is found to be above an accepted limit, or a new leachable is found, a safety evaluation will need to be performed. The chemist will need to provide the toxicologist with information that will help to qualify the leachable, including the identity of the leachable, which may include compound class or more specific information, such as chemical formula and structure; and the amount and concentration of the leachable in the drug product.
Information sharing between packaging component manufacturers/suppliers and drug product developers/manufacturers is also important in order to guide packaging component and materials of construction selection, provide knowledge of potential extractables and leachables, and facilitate leachables–extractables correlations via knowledge of packaging component chemical compositions, etc. (also see 1663).
LEACHABLES STUDY DESIGN
Although leachables studies may be accomplished at any time during the drug product development/manufacturing lifecycle, leachables studies are especially relevant during late stage product development or during formal product stability assessment. Ideally, leachables assessment is conducted as follows:
-
The assessment is performed on the actual drug product and not simulations thereof (however, see Simulation Studies).
-
The assessment is performed with the actual packaging and delivery system in the form it will be commercialized, not with a prototype or on system components.
-
The related extractables assessments are accomplished on the same lots of packaging components used to manufacture the drug product lots on which the leachables assessments are performed.
-
The assessment is performed on a product that is manufactured under conditions that reflect the actual commercial processes of production of the drug product and the packaging/delivery system, filling of the drug product into the packaging/delivery system, post-filling treatment of the filled packaging (e.g., terminal sterilization), distribution, storage, and clinical use of the drug product. Although leachables studies may include accelerated storage conditions, they cannot be limited to accelerated conditions and must include real-time assessment.
Leachables studies can also be performed early in the drug product development process (e.g., preclinical stage) in order to facilitate the selection of packaging components and their materials of construction. Such leachables studies are particularly useful for certain “high-risk” dosage forms (see Table 1) where selecting appropriate packaging components and materials of construction is critical. A variety of packaging components and materials of construction can be evaluated at the same time and drug product leachables profiles determined and evaluated for each configuration. For primary packaging systems or combination drug/device products this can be accomplished by using either the drug product formulation or a placebo formulation in contact with the proposed packaging system. In the latter case, the placebo formulation can be considered as a simulating solvent to characterize extractables as probable leachables (see Simulation Studies). In either case, the leachables study conditions (i.e., time, temperature, etc.) should be based on conditions that are relevant for either the use-life or shelf-life of the drug product. Preclinical development stage leachables studies can be designed in a systematic way in order to support QbD processes and principles. It is important to also note that during early stage drug product development for high-risk dosage forms, leachables characterization is recommended for any drug product batches that are used as test articles in any definitive toxicology or clinical studies. For “low-risk” dosage forms (e.g., solid orals, topical powders) leachables studies conducted throughout development might be appropriate in order to assess, and thereby avoid, problems with packaging systems that might appear either in later stage development or marketed product.
During later stage development of high-risk dosage forms in support of product registration, when the final market form of the packaged drug product is available, leachables studies may be accomplished on definitive registration batches of drug product during the course of overall product stability studies. The results of these leachables stability studies can be used to establish leachables–extractables correlations, identify trends in leachables accumulation levels, evaluate individual leachables and qualify them on a safety basis, and develop leachables specifications with acceptance criteria (should these be required). For inhalation aerosols and other OINDP, leachables testing should be an integral part of the larger ICH registration stability program (2), and storage conditions and stability time points should be planned accordingly. For cases where a packaging/delivery component is in direct contact with the patient (e.g., a metered dose inhaler or dry powder inhaler actuator mouthpiece), chemical entities that a patient might be exposed to can be evaluated as extractables (i.e., potential leachables) using appropriate simulating fluids under time/temperature exposure conditions relevant to the intended use (see 1663).
Additionally leachables assessments may be appropriate on certain occasions post-market. For example, drug product leachables studies may also be appropriate in many cases where necessary or desired changes in a marketed drug product are made. Such leachables studies are normally required to support change-control processes for many high-risk dosage forms, particularly those with in-place leachables specifications and acceptance criteria, and could also be appropriate for other dosage form types, drug/device combination products, etc. Changes may include but are not limited to: composition of the drug formulation; manufacturing processes for the drug product; primary and secondary packaging components or their materials of construction; manufacturing or assembly processes for primary and secondary packaging components or their materials of construction; and delivery system(s) that are part of the drug product labeling. Any change that results in the patient being exposed to a different leachables profile than the one approved during registration will require leachables studies as part of any change-control process unless adequate scientific justification is provided to the contrary.
Although low-risk dosage forms (e.g., solid orals, topical powders) typically do not rigorously require leachables studies as part of the drug product registration process (Table 1), it is possible that leachables could appear in drug product impurity profiles either during registration stability studies or in marketed products. For example, it has been documented that chemical additives in label adhesives can migrate through plastic primary packaging and appear in impurity profiles of solid oral dosage forms packaged within these containers. Thus, it is appropriate to consider performing leachables studies on “low-risk” dosage forms in certain cases. If leachables assessment is not performed proactively, such an event could lead to an OOS result for a development or marketed product and require an “emergency” leachables study as part of an investigation process. The design of this type of leachables study depends on the particular situation; however, in general it would be necessary to identify and quantify the leachable(s), evaluate safety and possibly qualify the leachable(s), and correlate the leachable(s) with packaging component extractables. It is also possible that leachables could result from contact with manufacturing equipment and tertiary packaging systems (e.g., shipping materials).
The design of a particular leachables study depends on the purpose and goals of the overall leachables assessment. Although the leachables studies described above have different purposes and overall goals, they require similar types of information for their proper design. First, it is important to have information as to the identities and maximum possible accumulation levels of all potential leachables. The packaging component manufacturers may provide chemical composition details for the packaging/delivery system and various materials of construction, as well as details regarding the manufacturing processes for these components and materials. Such information may be in the form of material safety data sheets, technical data sheets, test reports, or confidential communications, and can be used to infer potential leachables. An extractables assessment (including an extraction study) can also be accomplished on packaging components and/or their materials of construction to directly assess potential leachables (see 1663). Regardless of how the chemical information is obtained, it is important to ensure that all possible sources of potential leachables from the finished packaging system are considered. These may include chemical entities from any of the primary and secondary packaging components and their materials of construction, coatings, cleaning, lubricating, cutting, sterilization, assembly, or other processes associated with the manufacture of the final packaging/delivery system as used in the drug product. The chemical information on the packaging and delivery system is used to create a list of potential leachables and their possible accumulation levels.
Potential leachables have a significant chemical diversity, and therefore a diversity of physical and chemical properties, including polarity, volatility, solubility, etc. Whereas relatively volatile compounds can more readily migrate into any type of formulation through indirect contact, nonvolatiles generally require direct contact. Two aspects of formulation contact should be considered: the nature of the formulation contact (i.e., direct or indirect) and the time of contact (transient or continuous). If the formulation is not in direct contact with the packaging component (e.g., inhalation powder in a capsule packaged in a blister) then it is less likely that any relatively nonvolatile compounds would migrate into the formulation from the packaging system; however, volatile compounds might. If the formulation is only briefly contacting the packaging component (e.g., an inhaler mouthpiece) it is less likely that any migration of chemicals from the component would occur on this transient timescale. However, if the formulation is in continuous contact with the packaging component (e.g., parenteral in a bag delivered through an administration set) then all types of compounds could potentially migrate into the formulation.
A rigorous leachables assessment considers leachables from sources other than primary packaging, such as necessary secondary packaging and, in certain situations, tertiary packaging. If the primary packaging consists of a semipermeable polymer (e.g., a low density polyethylene container), then potential leachables from labels, inks, adhesives, etc. that are used in the secondary packaging must be evaluated. Similarly, volatile compounds that are present in tertiary packaging (e.g., wooden pallets, cardboard boxes, plastic overwraps, etc.) could migrate into a formulation contained in such a plastic bottle. These migrants that are derived from tertiary packaging should be considered in the event that an unknown impurity is detected or suspected in the drug product.
Various characteristics of the drug product formulation must also be considered in designing any leachables study. For example, formulations are typically either solids or liquids, and it is well documented that physical state affects the leaching process. In the event that a formulation has a change in state during the course of production (e.g., lyophilization; liquid to solid) then the leachables study should be designed taking into consideration the time periods that the formulation is expected to be in each physical state. In the event that a formulation has a change in state during the course of use (e.g., nebulization of a liquid to vapor) then consideration should be given to both the leachables acquired during storage from the container of the liquid and those acquired during use of the prescribed delivery device. In addition, typically only the final packaged product is evaluated for leachables; however, there may be cases in which an intermediate (e.g., bulk capsule for an inhalation powder) is stored for long periods of time in different primary packaging (e.g., foil pouch) from which compounds may leach. If these compounds that migrate from bulk packaging persist through the drug product's manufacturing process and are entrained in the finished drug product, then they are properly treated as leachables.
The nature of any contact that the packaging and delivery system has with the patient must also be considered. If the contact is only surface contact, then the likelihood of direct chemical migration to the patient is much less than if the contact is with mucosa, tissue, bone, or dentin. The various contact categories are described in The Biocompatibility of Materials Used in Drug Containers, Medical Devices, and Implants 1031 or ISO 10993 (10).
LEACHABLES CHARACTERIZATION
The primary goal of any leachables study is leachables characterization; i.e., the discovery, identification, and quantitation of leachables present in a particular drug product. Analytical methods for leachables characterization are developed based on the nature of the drug product matrix, the identities and possible accumulation levels of potential leachables, and the required sensitivity based on an adopted leachables evaluation threshold and the capabilities of the analytical methods employed. Unlike a typical drug product impurity method where target analytes are related to the drug substance, leachables have a wide chemical diversity and can come from various sources in the packaging/delivery system. Leachables also have a wide range of possible accumulation levels in a drug product. Taken in total, these factors present a significant challenge for trace analysis, especially in the case of organic leachables identification. Under certain circumstances, this challenge can be mitigated by performing the process of potential leachables identification outside of the leachables assessment, for example via extractables assessment in simulated extractables studies (see Simulation Studies).
Before describing the processes, analytical techniques, and methods involved in leachables characterization, it is appropriate to state that the ultimate objective of thorough leachables characterization as defined above cannot be realized in all cases, even when state-of-the-art analytical chemistry is practiced with best available skill and diligence. It is a reality that there is no analytical technique or combination of analytical techniques that is capable of the discovery, identification, and quantitation of any and all organic and inorganic leachables. For example, authentic reference compounds for organic leachables may not be available in all cases for confirmation of identifications or for quantitative instrument calibration. Given these circumstances, the practical objective of leachables characterization must therefore be the discovery, identification, and quantitation of individual leachables present in a drug product above a predetermined level, or “threshold”, to a reasonable degree of scientific certainty and exercised with appropriate due diligence.
Analytical Thresholds
The starting action in leachables method development is to establish the level at which the method must perform at to accomplish the appropriate leachables characterization functions. This level is known as the analytical threshold. Minimally, an appropriate method must function at all levels greater than or equal to the analytical threshold. As discussed previously, such an analytical threshold can be based on various criteria, including safety considerations. An example of a safety-based threshold is the SCT as established for OINDP. In order to define the SCT in terms that facilitate laboratory analysis, it must be converted from units of exposure (i.e., µg/day) to units of concentration (e.g., µg/mL, µg/g, µg/canister, µg/vial, etc.). This is accomplished by considering the dose parameters for a given drug product per the drug product's label claim. The resulting analytically useful threshold is termed the Analytical Evaluation Threshold (AET) (2). Previously characterized target leachables will have known safety profiles and previously established leachables thresholds. In any event, thresholds can be used for the basis for analytical method development unless other product considerations, such as compatibility, dictate a lower level is necessary.
A general formula for converting the SCT (0.15 µg/day) to an AET is as follows:
+'&img=../../images/v38332/c1664_form1_pf395.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Further, for liquid dosage forms:
+'&img=../../images/v38332/c1664_form2_pf395.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Further, for solid dosage forms:
+'&img=../../images/v38332/c1664_form3_pf395.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
This AET establishes the level at which unknown leachables should be identified and quantified in a particular drug product, and can therefore be used as a basis for analytical method development.
Analytical Method Requirements
Analytical method requirements for leachables characterization are based on the determined AET (or an alternative valid threshold concept), and information on potential leachables obtained from extractables assessments of packaging components and materials, including information from component/material suppliers. Since leachables are typically a subset of extractables or chemically linked to extractables, it may be the case that analytical methods used for leachables characterization can be based on those used for extractables characterization (see 1663). Any analytical method for leachables that is used for drug product stability studies in support of product registration, establishing leachables–extractables correlations for high-risk dosage forms, or the development of leachables specifications and acceptance criteria must be subject to complete validation using industry-accepted validation practices.
Preparing the Drug Product for Analysis—Sample Preparation
Sample preparation for leachables characterization is a function of the chemical nature of the potential leachables, the chemical nature of the drug product sample matrix, and the analytical technique(s) to be applied. The drug product matrix can present a significant challenge for organic leachables characterization. Drug product matrices contain the active pharmaceutical ingredient and excipients, which are typically present at high levels relative to leachables (except in certain high potency drug products). Analytical methods for organic leachables usually incorporate sample preparation procedures to separate leachables from the drug product matrix and concentrate them for analysis. The exact details of sample preparation procedures are unique to the individual drug product and while it is impossible to anticipate every scenario, the following general statements can be made:
-
Aqueous dosage form (e.g., inhalation solutions, small and large volume parenterals, ophthalmic solutions, etc.) leachables can be recovered using liquid-liquid extraction with water immiscible organic solvents, such as dichloromethane, hexane or petroleum ether, etc. The pH of the aqueous sample can be manipulated (i.e., raised or lowered) in order to enhance extraction of weakly acidic or basic leachables, or reduce extraction problems caused by the relatively high concentration of active pharmaceutical ingredient and excipients. The resulting organic extract can be dried if required (e.g., with magnesium sulfate drying agent) and concentrated if required by techniques that remove the solvent, such as evaporation with a gentle stream of dry nitrogen, rotary evaporation, or a Kuderna-Danish concentrator, etc. Concentrated organic extracts can be analyzed directly by GC-based methods; however, for HPLC-based methods using aqueous mobile phases the organic extract can be reduced to dryness (or near dryness) and the resulting residue of leachables taken up in a water miscible solvent (e.g., acetonitrile, methanol, etc.). Volatile leachables (e.g., solvents) can be analyzed directly from aqueous drug product samples with GC combined with headspace sampling. Note that recoveries of certain leachables can be affected by extraction and extract concentration procedures.
-
Solid dosage form (e.g., solid orals, inhalation powders, lyophilized powders, etc.) leachables can be recovered (for example) by dissolving the drug product with an aqueous solution and applying liquid-liquid extraction and extract concentration, as above. Headspace sampling and GC analysis of volatile leachables can be accomplished on the aqueous samples or, in some cases, directly on the solid dosage form. It is also possible to dissolve the drug product sample in another appropriate and analytically expedient medium (e.g., an organic solvent) for direct analysis by GC; however it is possible that matrix effects and interferences from the active ingredients and excipients could result.
-
Oral liquid dosage form leachables can be recovered by diluting the drug product sample in aqueous solution and applying liquid-liquid extraction and extract concentration, as above. Headspace sampling and GC analysis of volatile leachables can be accomplished on the aqueous samples or, in some cases, directly on the oral liquid dosage form. It is also possible to dissolve the drug product sample in another appropriate and analytically expedient medium (e.g., an organic solvent) for direct analysis by GC; however, it is possible that matrix effects and interferences from the active ingredients and excipients could result.
-
Cream and ointment dosage form leachables can be recovered by dissolving the drug product sample in an aqueous solution, filtering, and applying liquid-liquid extraction and extract concentration, as above. Headspace sampling and GC analysis of volatile leachables can be accomplished on the aqueous samples. It is also possible to dissolve the drug product sample in another appropriate and analytically expedient medium (e.g., an organic solvent) for direct analysis by GC; however, it is possible that matrix effects and interferences from the active ingredients and excipients could result.
-
Dosage forms with nonaqueous drug product vehicles (e.g., metered dose inhalers with organic solvent propellants) require special sample preparation procedures, which are discussed in 1664.1.
Development of sample preparation methods for leachables characterization can be accomplished through the use of appropriate test samples, such as accelerated drug product samples that have been aged under accelerated conditions (e.g., 40 /75%RH on 3-month storage), drug product or placebo samples spiked with known potential leachables, and/or a simulated drug product matrix spiked with known potential leachables. Recoveries of spiked potential leachables should be assessed and optimized during method development. Internal standards can be included to improve quantitative accuracy and precision.
/75%RH on 3-month storage), drug product or placebo samples spiked with known potential leachables, and/or a simulated drug product matrix spiked with known potential leachables. Recoveries of spiked potential leachables should be assessed and optimized during method development. Internal standards can be included to improve quantitative accuracy and precision.
Note that the sample preparation for leachables characterization must create a test sample in a form amenable to the analytical technique to be applied, and appropriately concentrated so that individual leachables can be characterized relative to the selected threshold.
Analytical Techniques
Analytical techniques applied to leachables characterization are the same as those applied to extractables characterization, which are summarized and discussed in 1663. Scouting analyses in general are not applied to leachables characterization, as the drug formulation may interfere with the scouting methods (see 1663). The most useful analytical techniques for discovery, identification (either by qualitative or structural analysis), and quantitation of organic leachables are those that combine GC and HPLC with mass spectrometry [i.e., GC/MS and HPLC (or LC)/MS]. Headspace sampling can also be interfaced with GC/MS to address volatile compounds. Other detection systems for both GC and HPLC that are not compound specific (e.g., FID, UV, etc.) are potentially useful for leachables discovery and quantitation, but not in general for identification. The combination of GC and HPLC techniques has the sensitivity and specificity required to characterize the diversity of chemical compounds found in many leachables samples. Analytical methods for leachables should be capable of characterizing target as well as new (or unspecified) leachables (e.g., scanning GC/MS or LC/MS); however, when additional sensitivity is required due to the use of analytically challenging thresholds, dedicated target compound methods (e.g., GC/MS with selected ion monitoring) can be used. It is also possible, with appropriate validation, to use methods based on techniques that are not compound specific (i.e., GC/FID, HPLC/UV, etc.).
Structural analysis of leachables should be accomplished with a systematic process identical to that described in 1663 for extractables, and to a level of confidence sufficient for safety assessment. A discussion of the principles of both gas and liquid chromatography is available in Chromatography 621. A discussion of the principles of mass spectrometry (including both GC/MS and LC/MS) is available in Mass Spectrometry 736. Although chromatographic-based hybrid analytical techniques are most commonly applied to leachables characterization, other analytical techniques with compound-specific detection capability (e.g., nuclear magnetic resonance spectroscopy) can be employed.
Quantitative Methods—Validation Considerations
Validation of quantitative leachables methods should be accomplished according to industry accepted practices, criteria, and standards, such as Validation of Compendial Procedures 1225 and (11). The extent of validation required depends on the goals of the leachables study in which the analytical method is being utilized. Validation parameters may include: accuracy, precision (repeatability, intermediate precision), specificity, limit-of-detection, limit-of-quantitation, linearity and range, and robustness. System suitability tests and criteria should also be developed for each leachables method. Special considerations for individual validation parameters relative to leachables methods are as follows:
-
Accuracy and precision—The validation parameters of accuracy and precision (repeatability and intermediate precision) are typically evaluated using drug product samples spiked with known amounts of target leachables. The choice of drug product spiking matrix used for these evaluations should be one that has had little-to-no contact with the packaging materials used in the final drug product, and therefore little-to-no measurable levels of endogenous leachables. Suitable spiking matrices can include freshly manufactured drug product and simulated drug product vehicles. Spiking levels should be determined based on results from accelerated stability studies or estimated from the known amounts of potential target leachables determined from extraction studies. Accuracy is typically performed at three spiking levels, which can also be determined based on results from accelerated stability studies or estimated from the known amounts of potential target leachables determined from extraction studies.
-
Linearity and range—Since potential leachables are present in packaging components at widely varying levels, actual drug product leachables can likewise appear at widely varying levels. The best accuracy and precision are achieved when the validated linear range considers the potential maximum accumulation levels of each target leachable or chemical class of leachables.
-
Limit of detection/Limit of quantitation—To detect and quantitate unknown leachables, the limit of quantitation should be at or below the designated analytical threshold (e.g., AET).
-
Specificity—Evaluation of method specificity can be accomplished by evaluating chromatographic peak purity in spiked and nonspiked drug product samples. For GC-based quantitative methods, this can be accomplished by GC/MS. For HPLC-based methods, either LC/MS or LC/DAD (diode array detection) can be used. Specificity can be qualitatively demonstrated if there are no observable method interferences related to the chemical entities present in the drug product.
-
Robustness—A design-of-experiments statistical approach with consideration of critical analytical method parameters (e.g., HPLC flow-rate, HPLC column, mobile phase gradient, etc.) should be used to create robustness evaluation protocols. Other approaches, such as serial change of critical parameters, can also be applied.
-
System suitability—Chromatography-based analytical methods, such as those described in 621, should include appropriate system suitability criteria for routine method evaluation, including tests for method linearity, precision, sensitivity, and specificity as appropriate. These parameters should be evaluated with an appropriately constituted test mixture(s) each time the quantitative leachables method is used, and should include appropriate system suitability acceptance criteria based on the method validation results. For example, sensitivity may be confirmed by analysis of standards prepared at the analytical threshold.
Several examples of validated leachables methods from the scientific literature have been documented in the chemical literature (12–17).
ESTABLISHING A LEACHABLES–EXTRACTABLES CORRELATION
A leachables–extractables correlation is established when actual drug product leachables can be linked either qualitatively or quantitatively with extractables from corresponding extractables assessments of individual materials of construction, packaging components, or packing systems. Leachables–extractables correlations are important for several reasons, including justifying the use of routine extractables release tests of packaging components as an alternative to leachables testing during stability studies for high-risk drug products, establishing the source of a leachable producing an OOS result for a low-risk drug product, change control, and ongoing quality control, etc.
A qualitative correlation is demonstrated when a leachable is linked either directly or indirectly to an extractable (i.e., potential leachable). For example, hexadecanoic acid observed in a leachables profile can be directly linked with hexadecanoic acid observed in the extractables profiles of one or more primary packaging components. The ethyl ester of hexadecanoic acid observed in the same leachables profile can be indirectly linked with hexadecanoic acid observed in one or more extractables profiles, if ethanol is a known drug product formulation constituent and it is shown that an esterification reaction can occur in the drug product during storage. For an appropriate quantitative leachables–extractables correlation to exist, the quantity of any individual leachable over the shelf-life of a drug product must be mathematically related to the quantity of the corresponding extractable in its source. One of the more simple mathematical relationships between an extractable and a leachable is that the quantity of the leachable in the drug product should be less than or equal to the quantity of the corresponding extractable. For example, the concentration of butylatedhydroxytoluene (BHT) present in a drug product formulation was determined to be 5 µg/mL. BHT was extracted from a primary packaging system component at a level of 300 µg/component. If the drug product packaging system incorporates one of these components per dosage form and the packaged formulation has a volume of 50 mL, then a quantitative leachables–extractables correlation is established, as BHT was extracted in the amount of 300 µg (300 µg/component × 1 component) and was leached in the amount of 250 µg (5 µg/mL × 50 mL). As a result, it can be concluded that on the average 50 µg of BHT is unaccounted for (300 µg extracted 250 µg leached), and that this quantity was either not leached from the packaging component into the formulation (more likely) or lost by some other process (less likely).
For high-risk drug products, leachables–extractables correlations may be established over multiple batches of drug product (accelerated or at end of shelf-life) and multiple batches of packaging components. Extractables studies should ideally be conducted on the same lots of components that were used to manufacture the drug product batches used in primary stability studies (and therefore on the drug product batches on which leachables testing was conducted to establish leachables–extractables correlations).
If the maximum level of any specific leachable in the formulation during stability studies was substantially greater than the calculated maximum potential accumulation levels of that same leachable as established by the extraction study, and the extraction studies were conducted on the same lots of components used to make the primary drug product stability batches, it can be concluded that the extraction study was incomplete and therefore a leachables–extractables correlation for that specific leachable cannot be established. In this case, either the extraction study can be augmented with experiments that produce an extractable level exceeding the maximum level of the leachable, or the leachable can be controlled via the drug product specifications for shelf-life stability testing, and release testing as an extractable at the component level is inadequate to control this leachable.
If a leachable–extractables correlation cannot be established, possible explanations include: inadequate extractables assessments of packaging components (see 1663); unreported changes in packaging component composition or manufacturing processes; unreported changes in the identity of packaging components.
CONSIDERATIONS IN DEVELOPING LEACHABLES SPECIFICATIONS AND ACCEPTANCE CRITERIA
The validated analytical methods and information obtained from those methods in a drug product leachables study can be used to develop drug product leachables specifications and acceptance criteria (i.e., limits). In certain circumstances, most commonly encountered with high-risk dosage forms (such as OINDP), it may be meaningful, useful, and at times required to routinely monitor finished drug products for leachables. Under such circumstances, leachables specifications and acceptance criteria must be established. One means by which such specifications and acceptance criteria could be developed includes testing a minimum of three drug product batches to determine their leachables levels. After thorough chemical and safety evaluation, the test data from the three or more batches can be used to establish acceptance criteria for targeted leachables, consistent with 1) the qualitative and/or quantitative results of leachables studies, 2) a consideration of the capabilities of the drug product's manufacturing process, and 3) a consideration of the potential safety, compatibility, and/or drug product quality impact of the leachables. It is important to note that leachables specifications should be applicable to a product during all stages of its shelf-life, including release and at end of shelf-life. This is the case since leachables accumulate over the entire shelf-life of a drug product.
When a change occurs in a product for which leachables specifications and acceptance criteria have been established, it is important to review the analytical method and re-evaluate the acceptance criteria and make adjustments to the specifications and acceptance criteria as appropriate and scientifically justified. A change in components that results in an increase in leachables concentrations beyond the levels qualified will necessitate the toxicological evaluation of the proposed levels, as would be the case for any impurity.
Acceptance criteria can be both qualitative and quantitative for both known and unspecified leachables. For example, a typical leachables specification could include:
-
Quantitative end of shelf-life limits for target leachables, which apply over the shelf-life of the drug product
-
A quantitative limit for “unspecified” (i.e., previously unidentified and uncorrelated) leachables (identification of unspecified leachables is required for accurate quantitation and toxicological evaluation).
ADDITIONAL CONSIDERATIONS
Simulation Studies
Occasions may arise in which it is not analytically feasible (due to challenging thresholds, for example) to successfully discover and identify all actual leachables in a drug product leachables study. This circumstance can be managed if the activities of discovery and identification of probable leachables are accomplished in an extraction study, where samples and analyte concentrations are more easily manipulated to achieve the necessary analytical performance. In such a circumstance, the actual drug product leachables assessment is simplified to a high-sensitivity quantitation of targeted leachables that have been discovered and identified as part of this extraction study.
In order to facilitate the discovery and identification of probable leachables, the extraction study must be similar in design to a drug product leachables study. Such an extraction study seeks to simulate the circumstances experienced by the drug product but should produce a test sample that is easier to characterize than the drug product itself. For such a study to be relevant in establishing appropriate target leachables, the solvent(s) used to generate the test sample must have nearly the identical propensity to leach as the drug product formulation. Such a study should be accelerated versus the leachables study so that the extractables, reflecting potential target leachables, can be discovered and identified in a timely fashion. Differences in the study design between this simulation study and the drug product leachables study are: 1) that the drug product formulation has been replaced with a simulating solvent that mimics the formulation; 2) that the conditions of contact have been accelerated, so as to increase both the concentrations of probable leachables and the rates of migration of probable leachables into the simulating solvent; and 3) that the test article can be either the complete packaging and delivery system or separate components of that system. Factors to consider in designing and justifying the simulating solvent(s), along with recommendations on the analytical approach used to characterize the simulating extract for extractables as potential target leachables are discussed in 1663. Given the intent of the simulation study, which is the discovery and identification of extractables as target leachables, simulation studies must also be driven by relevant thresholds.
It is possible that in cases of very low thresholds (e.g., AETs), quantitation of drug product leachables might still not be analytically feasible, even with high sensitivity target compound analytical methods. In such cases, the results of the simulation study (probable leachables identities and concentrations) may be sufficient to establish patient safety and the quality impact of the actual drug product leachables. To the extent that the simulation study mimics the drug product leachables study, the potential safety or quality impact of a compound as an extractable is an estimate of the potential safety or quality impact of the compound as an actual leachable. If it can be established that a compound quantitated as an extractable under these conditions has an acceptably small impact on safety and quality, then it follows that the same compound as a leachable in the drug product formulation may be assumed to have a similarly low impact on safety and quality as a leachable in the drug product formulation. The acceptability of this approach for any particular drug product needs to be scientifically justified by the drug product applicant.
If a compound is measured as an extractable in a simulation study and targeted as a leachable in a drug product leachables study, the extractables and leachables data for that compound become the basis upon which a leachables–extractables correlation can be made. For such a correlation to be considered to be valid, it is necessary that each leachable concentration in the drug product be less than or equal to the corresponding extractable concentration in the simulated extract, accounting for the uncertainty in the analytical measurements and any justifiable “exaggeration factors” that may have been utilized in the simulation study. Note that in certain justifiable circumstances drug product placebo batches may be used as test articles in drug product leachables studies (stability studies). However, there are also circumstances when placebo batches are not acceptable, such as when there is significant reason to believe that leachables might have an adverse effect on an active pharmaceutical ingredient (e.g., therapeutic proteins).
Inorganic (Elemental) Leachables
The topic of leachables as elemental impurities in pharmaceuticals can be addressed within the overall context of elemental impurities in drug products (e.g., Elemental Impurities—Limits 232. Elemental impurities leached from packaging or delivery systems represent only one source of elemental impurities in a drug product, and thus testing a drug product for elemental impurities does not establish that the impurities are leachables.
Testing of the plastic packaging systems and their materials of construction will establish those extractable elemental impurities that are relevant to a particular packaging system, and it may be appropriate to quantify such elemental impurities as leachables in the drug product. Therefore, the results of testing plastic packaging systems should be used to establish those elemental impurities that should be monitored as targeted elemental leachables in the drug product.
In general, guidelines and recommendations about elemental impurities in drug products address safety concerns associated with the elemental impurities. However, it is proper to consider elemental leachables from the broader perspective of the overall quality of the drug product. Thus the process of evaluating elemental leachables may include both the aspects of user safety and product quality.
Note that one of the differences between the testing for organic leachables (and extractables) and for elemental impurities is the nature of the information generated. The testing for organic leachables is based on having established the identity of the chemical compound that is the leachable. Alternatively, the test methods most commonly employed to address elemental impurities (atomic spectroscopy) do not establish the compound, or form, that the detected element is present in. For example, sulfur might be present in the form of elemental sulfur (S8), as the sulfate (SO42), or as a sulfur-containing organic compound (e.g., 2-mercaptobenzothiazole). Also, silicon might be present either as silicone oil or as silicon dioxide (SiO2). As the form of the elemental impurity may have a marked effect on the impurity's impact on product quality or safety, it may be the case that testing beyond elemental impurity profiling may be necessary to establish the exact chemical form of the elemental impurity and thus ascertain its potential safety or product impact.
An important issue to consider in testing drug products for elemental leachables is establishing which elements to measure as leachables. Considering this, it is noted that testing of the plastic packaging systems and their materials of construction will establish those extractable elemental impurities that are relevant to a specific packaging system. Therefore, the results of testing plastic packaging systems should be used as one means of establishing those elemental impurities that should be monitored as targeted elemental leachables.
SUMMARY
The requirement for, and completeness of, a leachables assessment for any particular drug product can only be determined by the drug product applicant with reference to appropriate regulatory guidance documents. Detailed recommendations for OINDP are presented in 1664.1.
Reference is also made to other compendial chapters in this Pharmacopeia that describe related extraction studies:
REFERENCES
-
FDA. Guidance for industry: container–closure systems for packaging human drugs and biologics. Rockville, MD: FDA; 1999.
-
Ball D, Norwood D, Stults C, Nagao L, eds. Leachables and Extractables Handbook. New York: J. Wiley and Sons; 2012.
-
Schroeder A. Leachables and extractables in OINDP: an FDA perspective. Paper presented at: PQRI Leachables and Extractables Workshop; 5–6 December 2008; Bethesda, MD.
-
Poochikian G. Leachables and extractables: evolution of regulatory aspects and perspectives on PQRI recommendations. Paper presented at: IPAC–RS 2011 Conference: Bringing Value to the Patient in a Changing World; 31 March 2011; Rockville, MD.
-
International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Q3B (R2): Impurities in New Drug Products, ICH Harmonized Tripartite Guideline, 2006.
-
Kroes R, Renwick A, Cheeseman M, Kleiner J, Mangelsdorf I, Piersma A, Schilter B, Schlatter J, van Schothorst F, Vos J, Würtzen G. Structure-based threshold of toxicological concern (TTC): Guidance for application to substances present at low levels in the diet. Food. Chem. Toxicol. 2004;42:65.
-
European Medicines Agency. CPMP/SWP/5199/02. Committee for Medicinal Products for Human Use. Guideline on the limits of genotoxic impurities; 2006.
-
PQRI Leachables and Extractables Working Group. Safety thresholds and best practices for extractables and leachables in orally inhaled and nasal drug products; 2006. http://www.pqri.org/pdfs/LE_Recommendations_to_FDA_09-29-06.pdf. Accessed 19 March 2013.
-
Ball D, Blanchard J, Jacobson-Kram D, McClellan R, McGovern T, Norwood D, Vogel M, Wolff R, Nagao L. Development of safety qualification thresholds and their use in orally inhaled and nasal drug product evaluation. Toxicol. Sci. 2007;97(2):226.
-
ISO 10993-18. Biological Evaluation of Medical Devices – Part 18: Chemical Characterization of Materials. International Organization for Standardization, 2005.
-
Swartz M, Krull I. Analytical Method Development and Validation. New York: Marcel Dekker; 1997.
-
Jenke D, Poss M, Story J, Odufu A, Zietlow D, Tsilipetros T. Development and validation of chromatographic methods for the identification and quantitation of organic compounds leached from laminated polyolefin material. J. Chromatogr. Sci. 2004;42:388.
-
Jenke D. Guidelines for the design, implementation and interpretation of validations for chromatographic methods used to quantitate leachables/extractables in pharmaceutical solutions. J. Liq. Chromatogr & Related Technol. 2004;27(20):1.
-
Jenke D, Garber M, Zietlow D. Validation of a liquid chromatographic method for quantitation of organic compounds leached from a plastic container into a pharmaceutical formulation. J. Liq. Chromatogr & Related Technol. 2005;28(2):199.
-
Xiao B, Gozo S, Herz L. Development and validation of HPLC methods for the determination of potential extractables from elastomeric stoppers in the presence of a complex surfactant vehicle used in the preparation of drug products. J. Pharm Biomed Anal. 2007;43:558.
-
Norwood D, Prime D, Downey B, Creasey J, Sethi S, Haywood P. Analysis of polycyclic aromatic hydrocarbons in metered dose inhaler drug formulations by isotope dilution gas chromatography/mass spectrometry. J. Pharm Biomed Anal. 1995;13(3):293.
-
Norwood D, Mullis J, Feinberg T, Davis L. N-nitrosamines as “Special Case” leachables in a metered dose inhaler drug product. PDA J. Pharm Sci Technol. 2009;63(4):307.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Desmond G. Hunt, Ph.D.
Senior Scientific Liaison (301) 816-8341 |
(GCPS2010) General Chapters - Packaging Storage and Distribution |
USP38–NF33 Supplement : No. 1 Page 7181
Pharmacopeial Forum: Volume No. 39(5)