



Add the following:
PURPOSE
This general information chapter presents a framework for the design, justification, and execution of an extractables assessment for pharmaceutical packaging and delivery systems. The chapter establishes critical dimensions of an extractables assessment and discusses practical and technical aspects of each dimension. Although intended to be helpful and generally applicable, the chapter is for informational purposes and does not establish specific extraction conditions, analytical procedures, or mandatory extractables specifications and acceptance criteria for particular packaging and delivery systems or dosage forms; nor does it delineate every situation in which an extractables assessment is required. It is not possible for a general discussion of extractables to anticipate and cover all situations where an extractables assessment might be required. Designing an individual extractables assessment is a process that balances sound science, prudent resource allocation, and effective risk management. Achieving this balance is the responsibility and obligation of the drug product manufacturer and assumes due consideration of all applicable legal and regulatory requirements. The principles and best demonstrated practices outlined in this general chapter represent a consensus interpretation of sound science and can therefore be applied to any situation in which an extractables assessment is required for pharmaceutical application.
KEY TERMS
This general chapter uses the following key terms listed below (1, 2; also see Packaging and Storage Requirements  659
659 ). Note that the terms Packaging System, Packaging Component, Primary Packaging Component, Secondary Packaging Component, and Materials of Construction are also defined in 659, and the definitions below are intended for clarification purposes within the context of this chapter and are not intended to supersede those provided in 659.
). Note that the terms Packaging System, Packaging Component, Primary Packaging Component, Secondary Packaging Component, and Materials of Construction are also defined in 659, and the definitions below are intended for clarification purposes within the context of this chapter and are not intended to supersede those provided in 659.
Packaging System (also referred to as a container–closure system): The sum of packaging components and materials that together contain and protect the product. This includes primary, secondary, and tertiary packaging components.
Delivery System: The sum of components and materials that are used to transport a drug product from its packaging to the point of administration into the patient. For example, an administration set is a delivery system that is used to transfer liquid drug products from their plastic packaging system to the site of administration to the patient. It is noted that in some cases, the packaging system itself may perform the delivery function.
Container: A receptacle that holds an intermediate compound, active pharmaceutical ingredient, excipient, or dosage form and is in direct contact with the product.
Closure: A material that seals an otherwise open space on a container and provides protection for the contents. It also provides access to the contents of the container.
Packaging Component is any single part of the package or container–closure system including the container (e.g., ampuls, prefilled syringes, vials, bottles); closures (e.g., screw caps, stoppers); ferrules and overseals; closure liners; inner seals; administration ports; overwraps; administration accessories; labels; cardboard boxes; and shrink wrap.
Primary Packaging Component is a packaging component that is in direct contact or may become in direct contact with the product (e.g., IV bag).
Secondary Packaging Component is a packaging component that is in direct contact with a primary packaging component and may provide additional protection for the product (e.g., overpouch or dustcover for an IV bag).
Tertiary Packaging is a packaging component that is in direct contact with a secondary packaging component and may provide additional protection of the product during transportation and/or storage (e.g., shipping carton for an overpouched IV bag).
Ancillary Component is a component or entity that may come into contact with a tertiary packaging component during the distribution, storage, and transportation of the packaged product (e.g., pallets, skids, shrink wrap).
Packaging Materials of Construction are substances used to manufacture packaging components. These are also referred to as Raw Materials.
Extractables are organic and inorganic chemical entities that are released from a pharmaceutical packaging/delivery system, packaging component, or packaging material of construction and into an extraction solvent under laboratory conditions. Depending on the specific purpose of the extraction study (discussed below), these laboratory conditions (e.g., solvent, temperature, stoichiometry, etc.) may accelerate or exaggerate the normal conditions of storage and use for a packaged dosage form. Extractables themselves, or substances derived from extractables, have the potential to leach into a drug product under normal conditions of storage and use and thus become leachables.
Leachables are foreign organic and inorganic chemical entities that are present in a packaged drug product because they have leached into the packaged drug product from a packaging/delivery system, packaging component, or packaging material of construction under normal conditions of storage and use or during accelerated drug product stability studies. Because leachables are derived from the packaging or delivery system, they are not related to either the drug product itself or its vehicle and ingredients. Leachables are present in a packaged drug product because of the direct action of the drug product on the source of the leachable. Thus leachables are typically derived from primary and secondary packaging, as the primary and secondary packaging can serve as a barrier between the packaged drug product and other potential sources of foreign chemical entities (such as tertiary packaging and ancillary components). In certain circumstances, packaging may directly contact the patient under typical clinical conditions of use (for example, the mouthpiece of a metered dose inhaler). As a result of this contact, patients may be exposed to leachables from the packaging without the action of the drug product. Leachables are typically a subset of extractables or are derived from extractables.
Migrants are also foreign organic and inorganic chemical entities that are present in a packaged drug product because they have leached into the packaged drug product from a packaging/delivery system, packaging component, or packaging material of construction under normal conditions of storage and use or during accelerated drug product stability studies. However, migrants are differentiated from leachables by the circumstance that migrants accumulate in the packaged drug product after the migrant has crossed a physical barrier, such as that provided by primary and secondary packaging. Because migrants cross a physical barrier, they are not present in the packaged drug product due to direct action of the drug product on the source of the migrant because the barrier prevents such direct action. Thus migrants are derived from secondary and tertiary packaging and ancillary components. Regardless of whether a substance is a leachable or a migrant, it is still a foreign substance in the packaged drug product and thus it must be impact assessed in the same manner. However, as the means by which a leachable and a migrant become entrained in a packaged drug product may be different, extractables studies meant to address leachables may be designed and implemented differently than extractables studies meant to address migrants.
Extraction Studies are the overall laboratory processes required in order to create extractables profile(s) of particular pharmaceutical packaging/delivery systems, packaging components, or materials of construction. Extraction studies are also referred to as Controlled Extraction Studies.
Characterization is the discovery, identification, and quantitation of each individual organic and inorganic chemical entity present in an extract above a specified level or threshold. Such thresholds can be based on patient safety considerations, materials considerations, the capabilities of analytical technology, etc.
Scouting is the process of acquiring general chemical information that provides insight into the nature and magnitude of extractables.
Discovery is the process of searching for, and ultimately finding, individual organic and inorganic chemical entities present in an extract.
Identification is the process of assigning a molecular structure to an organic extractable, or assigning constituent elements in the case of an inorganic extractable.
Quantitation is the process of measuring the level, or concentration, of an individual organic or inorganic chemical entity contained in an extract.
Extractables Profiles are qualitative and/or quantitative analytical representations of the extractables content of a particular extracting medium and set of laboratory extraction conditions.
Leachables–Extractables Correlations are established when observed drug product leachables are linked both qualitatively and quantitatively to extractables from associated packaging/delivery systems, packaging components, or materials of construction.
Safety Concern Threshold (SCT) is the threshold below which a leachable has a dose so low that it presents negligible safety concerns from carcinogenic and noncarcinogenic toxic effects.
Analytical Evaluation Threshold (AET) is the threshold at or above which a leachable should be characterized and reported for toxicological assessment. The AET can be mathematically derived from the SCT (or other threshold concepts) based on factors that include the dosing parameters of the drug product. When an extraction study is performed for the purpose of estimating the accumulation levels of leachables, the AET may be applicable to extractables as well as leachables. The concept of the AET is discussed in greater detail in Assessment of Drug Product Leachables Associated with Pharmaceutical Packaging/Delivery Systems 1664.
SCOPE
The scientific principles and best-practices described in this general chapter are intended to apply to any extractables assessment or extraction study of pharmaceutical packaging/delivery systems, packaging components, or materials of construction; the results of which are intended for establishing extractables profiles. Extractables profiles can be used in a variety of pharmaceutical development and manufacturing applications, including the characterization, selection, and qualification of materials of construction, packaging components or packaging/delivery systems; establishing leachables–extractables correlations; and/or simulating worst-case drug product leachables profiles. When appropriate, extractables profiles can also be used in establishing leachables–extractables correlations as described in Assessment of Drug Product Leachables Associated with Pharmaceutical Packaging/Delivery Systems 1664. These scientific principles and best practices can also be applied to materials of construction and functional components of equipment used to manufacture drug substances and drug products; e.g., filters, tubing, and tanks. In addition, these principles and best practices can be applied to materials of construction for the medical device component of a combination product (2), with appropriate consideration of the guidances and regulations that apply specifically to medical devices. These scientific principles and best practices apply to all organizations and individuals involved in the manufacture of drug substances, drug products, and in their stability studies, including but not limited to:
-
Manufacturers of drug substances and drug products for human and veterinary use where manufacturing may involve operations at the applicant holder's facilities (i.e., facilities that belong to the holder of an approved New Drug Application or Abbreviated New Drug Application) or at those of a contractor for the applicant holder
-
Manufacturers of combination products
-
Packaging operations by the manufacturer or a designated contractor for the application holder
-
Repackaging operations in which the drug product may be owned by an organization other than the primary manufacturer
Manufacturers and fabricators of pharmaceutical packaging/delivery systems, packaging components, and materials of construction may also apply these scientific principles and best-practices as appropriate.
BACKGROUND INFORMATION
During the course of manufacturing, packaging, storage, distribution, and administration; dosage forms and their constituents can contact components and materials of construction of manufacturing and packaging equipment, and primary and secondary packaging components and systems. Such contact may result in interactions between the dosage form and these components and materials. One such interaction is the migration or leaching of substances from any of these components and materials into the dosage form with subsequent delivery to the patient during drug administration. Patients also can be directly exposed to substances via direct contact with the packaging/delivery system during drug administration. Leachables, which can include both organic and inorganic chemical entities with wide chemical diversity, are of concern due to their potential safety risk to patients and potential compatibility risks for the drug product (e.g., drug substance interaction/degradation, pH change, appearance change, particle formation, protein aggregation/structure change, etc.). In order to assess these risks and manage the potential issues posed by leachables, it is necessary to know the identities and the levels to which leachables will accumulate in the finished drug product over its shelf-life. These two pieces of information can be used to establish the magnitude of patient exposure (dose) and therefore the safety risk posed by an individual leachable, as well as the likelihood of any compatibility issues involving the drug product.
Regulatory guidelines and various best-practice recommendations state that assessment of the potential impact of contact between a component or material and a final dosage form involves evaluating the final dosage form with respect to leachables. This assessment can include a migration or leachables study whose purpose is to discover, identify, and quantitate leachables that have migrated from the contacted system, components, or materials and accumulated in the dosage form. Alternatively, this assessment may involve performing a simulation extraction study, when use of such a study in lieu of a migration study can be justified. There are many science-based and practical reasons why such a leachables assessment typically does not stand alone as the single means of assessment. Since the pharmaceutical packaging/delivery system is the primary source of potential leachables, it is generally appropriate that any leachables assessment be preceded by an extractables assessment performed on the packaging/delivery system, its primary and certain critical secondary packaging components that are noncontacting but potentially interacting, and/or packaging and delivery system materials of construction; consistent with regulatory guidelines and best-practice recommendations. Such an extractables assessment can also be performed on particular components and/or materials of construction of manufacturing and packaging equipment, as well as certain tertiary packaging components, that are deemed of high leaching potential or have been implicated in an identified leachables problem with a particular drug product.
Extractables assessments can be used to:
-
Characterize packaging/delivery systems, packaging components, combination product medical device components, manufacturing components, and their various materials of construction
-
Facilitate the timely development of safe and effective dosage form packaging/delivery systems, manufacturing systems and processes by assisting in the selection of components and materials of construction
-
Understand the effects of various manufacturing processes (e.g., sterilization) on packaging components and their potential leachables
-
Establish the worst-case potential leachables profile in a manner which facilitates leachables studies, the development of leachables specifications and acceptance criteria (should these be required), and the safety evaluation/qualification of potential and actual leachables
-
Establish the worst-case potential leachables profile in a manner which facilitates the safety evaluation/qualification of probable leachables when it is not scientifically possible to determine actual leachables
-
Facilitate the assessment of patient exposure to chemical entities resulting from direct contact between a patient's body tissue(s) (e.g., mouth, nasal mucosa) and a packaging or combination product medical device component (e.g., a metered dose inhaler's plastic actuator/mouthpiece)
-
Facilitate the establishment of qualitative and quantitative leachables–extractables correlations
-
Facilitate the development of extractables specifications and acceptance criteria (if these are required) for packaging components, combination product medical device components, and materials of construction
-
Facilitate investigations into the origin(s) of identified leachables whose presence causes quality and/or safety issues (such as out-of-specification results) for a marketed product
In these ways, extractables assessments can support Quality by Design (QbD) principles for the development and manufacture of pharmaceutical packaging/delivery systems and drug products. Note that although characterization of packaging/delivery systems and materials is a goal of many extractables assessments, regulatory guidances and best-practice recommendations clearly stress that extractables assessments also serve as investigations into potential leachables (1–6).
As stated previously, it is not a goal of this chapter to identify each case in which an extractables assessment is required for packaging/delivery systems, individual packaging components, or materials of construction for any particular type of dosage form. This is the responsibility of the Holder of the NDA (applicant holder), and assumes appropriate consideration of applicable regulatory guidance documents. Rather, this chapter addresses the question “If an extractables assessment is required, what are the scientific principles and best demonstrated practices under which it should be accomplished?”
As performing an extractables assessment involves the processes of discovery and identification, an extractables assessment can be facilitated by knowledge about the test article, especially its composition. Thus it is strongly recommended that the test article assessor and the test article vendor collaborate in such a way that the test article assessor has access to critical information which will aid in the design and implementation of an effective and efficient extraction study. Additionally, it is noted that characterization of the test article per Plastic Materials of Construction 661.1 will provide information that is useful in the design and implementation of extraction studies.
Achieving the objectives of an extractables assessment requires performance of an extraction study in order to create extractables profiles. An extraction study has two critical dimensions: laboratory generation of the extract (extraction) and testing the extract (characterization).
GENERATING THE EXTRACT
General Concepts and Critical Experimental Design Parameters
Extractables are derived from a variety of sources and exhibit extensive chemical diversity. Primary sources of extractables include:
-
Chemical additives in individual elastomeric/polymeric packaging components and raw materials, including impurities in these additives
-
Chemical entities and additives that are present in packaging components composed of glass and metals
-
Entities related to the dissolution of the packaging component itself (e.g., iron extracted from a stainless steel material, silicon extracted from glass)
-
Monomers and higher molecular weight oligomers derived from incomplete polymerization
-
Migrants from secondary and tertiary packaging components, such as inks, label adhesives, and volatiles from cardboard shipping containers, plastic storage bags, and other shipping aids such as wooden pallets
-
Surface residues, such as heavy oils and degreasing agents on metal canisters and containers
-
Chemical substances on the surfaces of component fabrication machinery or other drug product manufacturing systems, such as mold release agents, and antistatic and antislip agents
-
Chemical additives, monomers/oligomers, impurities, etc., in various parts of component fabrication machinery or other drug product manufacturing systems
As noted above, the chemical diversity of extractables is significant. For example, the chemical additive category antioxidants includes hindered phenols, secondary aromatic amines, hindered amines, organosulfur compounds, organophosphorus compounds, and other chemical classes.
Extraction is a process of treating a material with a solvent to remove soluble substances. Extraction is a complex process influenced by time, temperature, surface area to volume ratio (i.e., stoichiometry), extraction medium, and the phase equilibrium of the material (3). There are two reasons why an extraction study is the necessary and appropriate means of accomplishing the various objectives of an extractables assessment. First, there is no other viable analytical alternative. Characterizing a material for potential leachables in its natural solid state is a goal of modern analytical chemistry rather than an accomplishment. Second, even if a direct characterization could be accomplished, it would at best only establish the identities and levels of chemical entities present in the material, and not assess the leaching characteristics of these chemical entities. A compositional assessment does not take into account any chemical reactions that can alter the molecular structures of potential leachables over the dosage form's lifecycle. For example, in the case of phenolic antioxidants that are leached by an aqueous drug product, hydrolysis and oxidation products can accumulate in the drug product. The only viable means for producing data related to leaching is to use a process such as laboratory extraction that is mechanistically similar to leaching.
The design of an extraction study is dictated by the purpose of the extractables assessment and the question(s) being asked, as well as the available information regarding the chemical composition of the test article(s) to be extracted. Extraction studies can be designed to answer questions such as:
-
What are the chemical additives in a particular packaging component or material of construction?
-
What are the maximum accumulations of chemical additives from a particular packaging component into the dosage form?
-
What are the likely contents of an end-of-shelf-life drug product leachables profile?
Addressing each of these questions requires a particular set of parameters, such as extraction time, extraction temperature, extracting solvent, extraction technique, sample surface area to extracting solvent volume ratio, etc. Clearly, the intent of the extractables assessment must be established before the study design is finalized. For example, if the purpose of the extraction study is to simulate a worst-case leachables profile, then the study can be termed a simulation study that should produce an extract that:
-
Contains all the substances (extractables) that could leach into the final product at levels considered potentially significant
-
Contains these extractables at concentrations that are greater than or equal to the maximum concentration that these chemical entities will accumulate in the drug product as leachables at any time during the shelf-life
-
Is generated more efficiently and in less time than that required for a drug product leachables study
-
Is amenable to chemical analysis
The concept of a simulation study is addressed in greater detail in Assessment of Drug Product Leachables Associated with Pharmaceutical Packaging/Delivery Systems 1664. The means by which an extraction process is accomplished are reflected in the juxtaposition of several experimental parameters including:
-
The chemical nature of the extracting media
-
The time duration of the extraction process
-
The temperature and pressure at which the extraction is performed
-
The stoichiometry of the extraction process (extracted surface area per unit volume of extracting solution)
-
The mechanism or process by which the extraction is accomplished
Extraction processes have been described as “accelerated”, “aggressive”, “exhaustive”, “vigorous”, “harsh”, and so on, and for medical device studies certain of these terms have been defined (7). In general, extraction processes should allow completion in a reasonable time frame but should not be so aggressive that they alter the qualitative and/or quantitative nature of the resulting extractables profile. The most aggressive extraction conditions are reserved for the quantitative determination of chemical additive contents in components and materials. Because such studies are intended to quantitate specific known chemical additives and not to simulate a drug product leachables profile, it is acceptable to use extraction conditions which disrupt or dissolve the component or material being extracted, and thus to alter the resulting extractables profile, while recovering the target additive(s) without loss or chemical decomposition.
Chemical Nature of the Extracting Medium
Of all the parameters involved in generating the extract, the extracting medium is the most critical because it is the extracting medium that accomplishes the extraction, and all other parameters merely facilitate the extraction. Establishing and justifying the extracting medium (or media) is both straightforward strategically and complex tactically. Strategically, if the purpose of a particular extraction study is, for example, to simulate a worst-case leachables profile, then the ideal situation is for the extracting solvent to have a similar or greater propensity to extract substances as the formulation, thus obtaining a similar qualitative and quantitative extractables profile. This is clearly stated in regulatory guidances and best practice recommendations (1, 4–6). Therefore, the most logical tactic for this simulation study is to use the formulation itself as the extracting medium and in the absence of complicating factors, such an approach is recommended. However, in certain cases the use of the formulation as an extracting medium complicates extract characterization to such an extent that it is impractical. The various guidances and recommendations suggest that if the use of the drug product as the extracting solvent is not feasible, then the drug product vehicle, or placebo, could be used as an effective extracting medium. This recommendation is derived from the fact that the drug substance itself does not typically create the “leaching power” of a drug product but rather that it is the formulation's ingredients (drug product vehicle) that establish the drug product's ability to leach substances from a contacted material.
When circumstances require that an extraction study must be accomplished with a simulating solvent(s), it is necessary to establish and justify the composition of these solvents. In order to accomplish this objective, one must consider all the physicochemical characteristics of a formulation and/or simulating solvent that influence its “extracting power”. In certain circumstances, the formulation is sufficiently simple that the critical characteristics can be readily delineated and simulated. For example, the extracting power of polar aqueous drug products consisting of soluble ingredients (such as an injectable with a drug substance, buffers, and diluent) is, for organic extractables, driven primarily by drug product pH. In such a circumstance, simulating the drug product pH with an analytically viable buffer system for the extraction study may be appropriate and justifiable. For inorganic extractables, utilization of a simulating solvent having similar metal-chelating properties as the drug product vehicle may also be appropriate and justifiable. It may also be the case that largely non-polar drug products can be readily simulated with analytically expedient organic solvents. For example, chlorofluorocarbon and hydrofluoroalkane propellants used in metered dose inhalers (MDIs) can be simulated with dichloromethane as an extracting solvent and isopropanol can be used to simulate ethanol, a common co-solvent in MDI formulations.
Many drug products are compositionally intermediate between the polar and nonpolar examples just discussed. Examples of such products include “aqueous” drug products that contain stabilizers, solubilizing agents, chelating agents and buffers, lipid-containing products, and biotechnology products containing proteins, peptides, and blood-derived products. Such products have a characteristic polarity which establishes their “extracting power”. Thus, an appropriate simulating solvent will have a polarity that matches that of the drug product. Binary mixtures of miscible solvents (such as alcohol/water) have been utilized as simulating solvents for these types of drugs product.
It may be that a single simulating solvent cannot be established and justified for a specific drug product, or that the focus of the extractables assessment is a material or system that will be characterized for use with multiple, compositionally diverse drug products. In such circumstances, the drug product's ability to leach chemical entities from a packaging system can be established based on the use of multiple extracting solvents, each of which addresses one (or more) of the extracting “mechanisms” that are relevant to the drug product (or drug products) under investigation. The use of multiple solvents is consistent with industry-driven best-practice recommendations for drug products that have a relatively high risk of dosage form interaction with the packaging system and a relatively high safety risk relative to the route of administration (e.g., inhalation aerosols and solutions, injectables and injectable suspensions) (1). Therefore, the use of multiple solvents (or extracting media) with different polarities, pH, ionic strength, or extracting powers, is recommended for high risk dosage form packaging system components and materials requiring extraction studies in order to simulate a drug product leachables profiles (see Table 1). If the goal of an extractables assessment is materials characterization, then simulating a drug product vehicle is both unnecessary and undesirable since this goal requires qualitatively and quantitatively efficient extractions. Such extractions generally are only achieved with relatively powerful organic solvent systems capable of softening, swelling, or dissolving the material's polymer matrix and releasing quantitative levels of additives and other chemical entities.
Table 1. Possible Extracting Media Relative to Particular Packaging Components
| Packaging Component |
Possible Extracting Mediaa
|
|---|---|
| MDI valve elastomer seal (MDI formulation contains 1,1,1,2-tetrafluoroethane and ethanol) |
Nonaqueous solvents (e.g., Dichloromethane Isopropanol Hexane)b
|
| Dry powder inhaler mouthpiece |
Water (unbuffered) Isopropanolc |
| Small-volume parenteral vial rubber stopper (aqueous formulation buffered at pH 6.5) |
Water (pH 5.2) Water (pH 9.5) Isopropanol:water (50:50)d |
| Large-volume parenteral plastic bag (aqueous formulation buffered at pH 7.2) |
Water (pH 5.2) Water (pH 9.5) Isopropanol:water (50:50)d |
|
a
The possibilities listed in Table 1 are provided for example only and should not be interpreted as standard practice recommendations.
b
These extraction media reflect the varying polarities of the organic solvents used in MDI formulations.
c
These extraction media reflect both the hydrophilic and lipophilic character of human saliva and allow materials characterization.
d
These extraction media reflect the chemical nature of the formulation. Using media whose pH range encompasses, and slightly exceeds, the pH limits of the product addresses the potential effect of pH on the extractables profile. The use of an aqueous mixture containing an organic solvent takes into account the possible presence of formulation additives such as solubilizing agents that can influence the leaching power of the formulation. The specific organic solvent used and its proportion in the extracting medium depends on the specific chemical nature of the formulation and on practical issues associated with testing the extract.
|
|
If the goal of an extractables assessment is materials characterization, then simulating a drug product vehicle is both unnecessary and undesirable since these goals require qualitatively and quantitatively efficient extractions. Such extractions generally are only achieved with relatively powerful organic solvent systems capable of softening, swelling, or dissolving the material's polymer matrix and releasing quantitative levels of additives and other chemical entities.
Extraction Time and Temperature
Extraction time and temperature are critical factors in the extraction process. Although the nature of the extraction solvent establishes the magnitude of the extraction (i.e., the amount of substances that can be extracted from a material at equilibrium), the combination of extraction time and temperature establishes the magnitude of the driving force and the degree to which equilibrium is actually achieved. In a simulating extraction study the purpose of elevated temperature is to increase the extraction rate, so that a short experimental time may simulate longer leaching times (1, 5–9).
Because extraction is a diffusion process, the relationship between the diffusion rate and temperature can be expressed empirically by the Arrhenius equation. The mathematics involved in a process that is driven by Arrhenius kinetics have been established in ASTM F1980-07 (2011) Standard Guide for Accelerated Aging of Sterile Barrier Systems for Medical Devices (10), which may be a useful guide for establishing accelerated contact conditions. As with all such models, the proper use of this model requires an understanding of the model's basis and essential principles, assumptions, and limitations (2).
Extractables profiles obtained with a given extracting medium and extraction technique can and should be monitored for equilibrium or the attainment of asymptotic levels of extractables (see Figure 1).
+'&img=../../images/v38332/c1663_fig1_pf395.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 1. A graphical representation of an extraction that has attained equilibrium as indicated by the achievement of asymptotic levels of target individual extractables as a function of extraction time (i.e., GC/MS peak area ratios of target extractables relative to an internal standard plotted versus extraction time).
Extraction Stoichiometry
Extraction stoichiometry considers the physical mass and/or surface area of the test article relative to the volume of the extracting medium, and the actual physical state of the material when it is extracted. Extraction stoichiometry can be manipulated to facilitate production of a more concentrated extract. For example, consider the case of a rubber stopper for a vial that contains 5 mL of a liquid drug product. A more concentrated extract than the drug product (i.e., an extract that contains higher levels of extractables than the leachables level in the drug product) could be produced by extracting 20 stoppers in 200 mL of extracting solvent. Another aspect of extraction stoichiometry is the physical state of the test article. It is not uncommon that components or materials are cut, opened, ground, or otherwise altered in size or configuration prior to being extracted. For inhomogeneous or layered materials such as film laminates, the process of cutting or grinding prior to extraction may alter the extractables profile as it may provide a means for the extracting solvent to come into contact with (and thus more effectively extract) materials (layers) that are shielded from contact with the solution under normal conditions of use. One can argue that the use of such sized material further facilitates the extraction process, however it is possible for sizing to reveal extractables that might not appear as leachables. Nevertheless, some sizing of components or materials before extraction can be useful in certain situations and for certain purposes, including: (a) reducing sample-to-sample variability by the consistent preparation of ground homogeneous polymeric material; (b) reducing the sizes of large packaging components to allow use of standard laboratory glassware for extraction studies; (c) increasing the surface area of a packaging component or material test article (e.g., via extruding, pressing, or grinding) in order to increase extraction efficiency. In any event, careful consideration should be given to the effect of physical sizing of test articles on the extractables profile before such sizing methods are employed in extraction studies.
For extractables assessments involving components or materials whose chemical ingredients are known based on information from the supplier or fabricator, analysts can manipulate extraction stoichiometry based on the known levels of chemical additives and the known sensitivity of the analytical technique(s) that will be used to characterize the extract. For example, consider the formulation for a peroxide-cured ethylene-propylene-diene-monomer (i.e., EPDM) gasket from an MDI valve shown in Table 2.
Table 2. Ingredients in a Peroxide Cured Rubber Gasket Test Article that are Used in an MDI
| Elastomer Ingredient |
Amount (Nominal) |
|---|---|
| EPDM polymer |
64.0% |
| Mineral fillers (may include stearic acid) |
34.4% |
| Antioxidant 1: (butylated hydroxytoluene) |
0.3% |
| Antioxidant 2: (2,2¢-methylene-bis-[6-(1,1-dimethylethyl)-4-methyl] phenol) |
0.3% |
| Peroxide curing agent |
1.0% |
Such information, when available from component and material suppliers, can be useful in designing an extraction study.
Analysts can also base the extraction stoichiometry on established safety thresholds for leachables. For example, an exposure of 0.15 µg/day total daily intake for an individual organic leachable has been proposed as an SCT for inhalation drug products, also termed orally inhaled and nasal drug products (5). Leachables present at or above the SCT, in an MDI for example, should be analytically and toxicologically evaluated, suggesting that extractables assessment also should be guided with the SCT in mind. The application of thresholds such as the SCT and AET to leachables assessments is discussed in greater detail in Assessment of Drug Product Leachables Associated with Pharmaceutical Packaging/Delivery Systems 1664.
In summary, extraction stoichiometry (and thus the “sensitivity” of an extraction study) can be based on:
-
The known chemical ingredients in a component or material
-
Safety-based thresholds for drug product leachables
-
The known or determined sensitivities of analytical instrumentation used for extract characterization
Mechanism of Extraction—Extraction Technique
An extraction can be accomplished in a variety of ways. It is necessary that the means of performing the extraction match the objectives of the extractables assessment. Common laboratory extraction techniques include:
-
Maceration (solvent soaking)—in which the test article is allowed to soak for a period of time in an organic or aqueous extracting solvent at temperatures below the solvent's boiling point. Analysts can also fill packaging system units with extracting solvent and store them at relevant temperatures.
-
Reflux—in which the test article is immersed in boiling solvent for a period of time.
-
Soxhlet—in which the test article is placed in the “thimble” of a Soxhlet extraction apparatus that is slowly filled with redistilled solvent from a boiling flask/condenser system; and periodically, the extracting solvent (containing extractables) is siphoned back into the boiling flask and the process begins again (for as many times as required to attain equilibrium).
-
Sealed vessel—in which the test article and extracting solvent are sealed inside a container capable of withstanding elevated temperatures and pressures, placed into a laboratory autoclave and heated with steam for a period of time.
-
Instrument-based solvent extraction—in which the test article is placed inside a sealed apparatus and extracted in an automated cycle; examples include pressurized fluid extraction, microwave-assisted extraction, and supercritical fluid extraction.
-
Sonication—in which the test article and extracting solvent are placed into a glass container and partly immersed in water inside an ultrasonic bath.
Each of these extraction mechanisms/techniques has its own unique advantages and limitations. For example, reflux extraction is very efficient, but may be too harsh for certain applications and can lead to thermal decomposition of certain organic extractables; the extracting power of sonication can be difficult to control; and because of its relatively high boiling point, water performs poorly in reflux and Soxhlet but well in a sealed vessel.
If the goal of the extractables assessment is identification and quantitation of the chemical additive content of a component or material, it is typical to use extraction techniques and processes that soften, swell, or dissolve (or in the case of inorganic extractables, digest) the component or material, thereby releasing quantitative amounts of chemical additives for analysis.
Extractions That are Not Solvent Mediated
Not all drug product or material-contact situations are solution mediated and not all issues related to leaching of material-derived entities involve a solution phase. For example, doses of inhalation powder contained in a capsule or blister pack for use in a dry powder inhaler may have volatiles leached from the capsule or blister material, or by specialty surface additives such as mold-release agents; a solid oral dosage form could contain volatile leachables derived from the adhesive of a paper label affixed to the plastic bottle that contains the dosage form; and inhalation solutions packaged in low-density polyethylene containers could contain volatile migrants from tertiary packaging or auxiliary components such as wooden shipping pallets. In the latter two cases, chemical entities can migrate through the plastic containers and volatilize into the airspace, subsequently accumulating as migrants in the dosage forms.
Extraction techniques specifically designed for application to volatile organic compounds are usually directly coupled to analytical instruments. These extraction techniques include headspace analysis (as headspace gas chromatography; HD/GC), direct thermal desorption (usually coupled to gas chromatography; TD/GC), and thermogravimetric analysis (TGA/GC).
CHARACTERIZING THE EXTRACT
Objectives and Challenges
Once an extract has been generated, the next objective is to perform a thorough chemical characterization of the extract. Setting a threshold (as described above), which is a specified level of an individual extracted chemical entity which requires characterization, can be based on safety considerations such as the SCT; functional considerations including nominal levels of known chemical additives in the formulation of an extracted component or material; or technological considerations such as the known or determined sensitivity of an analytical technology, instrument, or method. The extract characterization phase of the extraction study must enable the realization of the overall goals of the extractables assessment.
The ultimate objective of thorough extract characterization as defined above cannot be realized in all cases, even when state-of-the-art analytical chemistry is practiced with best available skill and diligence. It is a reality that there is no analytical technique or combination of analytical techniques that is capable of the discovery, identification, and quantitation of any and all organic and inorganic extractable chemical entities known to science. In some cases, authentic reference compounds for organic extractables may not be available for confirmation of identifications, or for quantitative instrument calibration. Thus, the practical objective of extract characterization must therefore be an exercise of due diligence in the discovery, identification, and quantitation to a reasonable degree of scientific certainty of all individual extractable chemical entities present in an extract above a specified level or threshold.
Processes Involved in Extract Characterization
1. scouting
The most useful analytical techniques in a scouting exercise are not compound specific, as they do not provide chemical information specific to the molecular structure of any particular extractable or chemical class of extractables. These analytical techniques provide information regarding bulk chemical properties of organic and/or inorganic chemical entities present in an extract, which can be used to guide extractables discovery, identification, and quantitation. Scouting analysis is not capable in and of itself of realizing the practical objective of extract characterization, regardless of which scouting technique or combination of techniques is applied.
Analytical techniques which can be employed for scouting are listed in Table 3, along with the particular bulk chemical property (and potential utility of this property) available from each technique. Some examples of the utility of scouting include the following:
-
Significant levels of nonvolatile residue determined by gravimetric analysis could suggest the presence of significant levels of inorganic chemical entities in the extract. This suggestion would be reinforced if significant mass remained after ashing the extracted nonvolatile residue (residue on ignition).
-
Significant UV absorbance of an extract suggests that organic chemical entities are present which contain UV chromophores within their molecular structure, such as phenolic antioxidants.
-
Characteristic features in an infrared spectrum of this extract could provide more detailed insights into the chemical classes of organic extractables present. These insights could be used to develop and apply analytical methods for discovery and identification that would detect the chemical classes of extractables suggested by the scouting process.
-
For aqueous extracts, total organic carbon provides a measure of the total amount of organic extractables present.
The scouting process and scouting analyses are optional for extract characterization. The utility of scouting is in the guidance it potentially provides for discovery, identification, and quantitation.
Table 3. Survey of Analytical Methods for Extract Analysis
| Analytical Technique |
Analytical Method |
Application |
Information/Utility |
|||
|---|---|---|---|---|---|---|
| Scouting |
Discovery |
Identification |
Quantitation |
|||
| Spectroscopy |
UV |
X |
|
X |
|
Bulk property of UV absorbing organic extractables; semi-quantitative with limited identification ability |
| FTIRa
|
X |
|
X |
|
Bulk property of IR absorbing organic extractables, moderate identification ability |
|
| Wet Chemical |
NVRb, ROIc
|
X |
|
|
|
Bulk property reflecting total amount of nonvolatile organic and/or inorganic extractables |
| pH |
X |
|
|
|
Bulk property of acidic or basic extractables |
|
| TOCd
|
X |
|
|
|
Quantitative measure of organic extractables |
|
| Gas Chromatography |
FIDe
|
|
X |
X |
X |
Discovery and quantitative assessment of individual organic extractables; note that qualitative identification is possible |
| MS |
|
X |
X |
X |
Discovery, identification, and quantitation of individual organic extractables; note that identification can be either qualitative or structural |
|
| FTIRa
|
|
X |
X |
|
Discovery and identification of individual organic extractables; note that FTIR has limitations relative to structural analysis (however identification via qualitative analysis is possible) |
|
| Liquid Chromatography |
UV, CADf, and ELSDg
|
|
X |
|
X |
Discovery and quantitative assessment of individual organic extractables; note that identification via qualitative analysis is possible and that Diode Array UV detectors can assist with structural analysis |
| MS |
|
X |
X |
X |
Discovery, identification, and quantitation of individual organic extractables; note that identification can be by either qualitative or structural and that ionization sources with different selectivities are available |
|
| FTIRa
|
|
X |
X |
|
Discovery and identification of individual organic extractables; note that FTIR has limitations relative to structural analysis (however identification via qualitative analysis is possible) |
|
| NMRh
|
|
X |
X |
|
Identification of individual organic extractables; note that identification can be by either qualitative or structural |
|
| Ion Chromatography |
Conductivity |
|
X |
|
X |
Discovery and quantitation typically of individual ionic species |
| MS |
|
X |
X |
X |
Discovery, identification, and quantitation of individual ionic extractables; note that identification can be by either qualitative or structural and that ionization sources with different selectivities are available |
|
| Spectrometry |
MS |
|
|
X |
|
Identification of individual organic extractables |
| NMRh
|
|
|
X |
|
Identification of individual organic extractables |
|
| IMSi
|
|
X |
X |
X |
Discovery and quantitative assessment of individual organic extractables; note that various ionization sources are available and that qualitative identification is possible |
|
| Atomic Spectroscopy |
AASj
|
|
X |
X |
X |
Discovery, identification, and quantitation of individual extracted elements (trace elements, metals); note that AAS can be applied to only one element at a time. Identification of the chemical form or speciation of the extracted element may require additional testing |
| ICP-AESk
|
|
X |
X |
X |
||
| ICP/MSl
|
|
X |
X |
X |
||
|
a
FTIR = Fourier Transform Infrared spectroscopy.
b
NVR = Nonvolatile Residue.
c
ROI = Residue on Ignition.
d
TOC = Total Organic Carbon.
e
FID = Flame Ionization Detection. Additional GC detectors, such as Thermal Energy Analysis Detector (TEA), may provide greater sensitivity for specific compound classes.
f
CAD = Charged Aerosol Detector.
g
ELSD = Evaporative Light Scattering Detector.
h
NMR = Nuclear Magnetic Resonance spectroscopy.
i
IMS = Ion Mobility Spectrometry.
j
AAS = Atomic Absorption Spectroscopy.
k
ICP-AES = Inductively Coupled Plasma Atomic Emission Spectroscopy.
l
ICP/MS = Inductively Coupled Plasma Mass Spectrometry.
|
||||||
2. discovery
The process of discovery involves testing an extract and thereby producing one or more analytical results that are attributable to individual extractables. The process of discovery is accomplished by detecting instrumental responses from the individual organic and inorganic extractables that are proportional to the levels of these individual extractables within the extract. It is in the discovery process that analytical techniques typically associated with trace organic and inorganic analysis are first required for extract characterization. Trace organic analysis typically involves the use of chromatographic techniques, particularly gas chromatography (GC) and high-performance liquid chromatography (HPLC). GC has enormous separating capability for volatile and semi-volatile organic compounds while HPLC is most applicable to semi-volatile and relatively nonvolatile organic compounds, making the two separation techniques complementary and orthogonal for application to the significant chemical diversity of extractables. A discussion of the principles of both gas and liquid chromatography is available in Chromatography 621.
The chemical diversity of extractables with respect to polarity and volatility can require alternative sample introduction techniques or sample modification, particularly for GC. Relatively volatile extractables like methanol are most amenable to headspace sampling of aqueous-based extracts into a GC. Organic acids and bases can often be analyzed more effectively by GC after chemical derivatization, such as methylation or silylation for organic acids. Both GC and HPLC can employ detection systems with different specificities (Table 3) which take advantage of unique structural properties of various chemical classes of extractables.
The analytical techniques useful for organic extractables discovery can also be applied to identification as well as quantitation. Analytical techniques such as gas chromatography/mass spectrometry (GC/MS), that are most often applied to identification, can also be used for both discovery and quantitation (Table 3). Inorganic extractables such as trace elements and metals are typically discovered, identified, and quantitated by the same suite of analytical techniques, such as atomic emission spectroscopy. Analytical techniques designed to study inorganic speciation, particularly in aqueous extracts, are considered beyond the scope of this chapter.
It is important to state that the overall goals of an extraction study always require the identities and quantitative amounts of individual organic and inorganic extractables, and so the mere discovery of extractables does not achieve the ultimate objectives of an extraction study.
3.identification
Identification of an extractable can be accomplished either by structural analysis or qualitative analysis. Structural analysis is the process by which the molecular structure of an unknown analyte is elucidated from compound-specific data, and therefore requires compound-specific detection of the unknown analyte. A compound-specific detector is one that provides information specific to the molecular structure of the individual unknown analyte (not just its chemical class). Qualitative analysis is the process by which an unknown analyte is matched with an authentic reference compound via one or more analytical techniques. The analytical techniques used for qualitative analysis can, but do not need to be, compound specific.
The analytical techniques most applicable to structural analysis, and to trace organic analysis problems in general, involve the combination of chromatography with mass spectrometry. These are the so-called “hyphenated” techniques of GC/MS and high-performance liquid chromatography/mass spectrometry (LC/MS). A discussion of the principles of mass spectrometry (including both GC/MS and LC/MS) is available in Mass Spectrometry 736.
Both GC/MS and LC/MS are capable of generating extractables profiles in the form of chromatograms. However, since LC/MS includes a relatively high chemical background of HPLC mobile phase ions, it is typical to include a non-destructive UV detector in series with the mass spectrometer to assist in locating peaks of individual extractables. The compound-specific data available from mass spectrometry include:
-
The monoisotopic molecular weight of the extractable based on confirmation of the molecular ion from one or more ionization processes
-
The molecular formula of the extractable based on accurate mass measurements, and/or accurate isotope ratio measurements, of the molecular ion
-
The fragmentation behavior of the extractable based on in-source fragmentation or tandem mass spectrometry
GC/MS interfaced with electron ionization produces mass spectra which can be searched through computerized databases, or libraries, of mass spectra from known compounds. Note that searchable mass spectra are generally unavailable for LC/MS ionization processes because of the variable nature of such spectra over time and between various instruments and laboratories. Both GC/MS and LC/MS also include the retention time (or retention index) of the unknown extractable which can be compared with that of authentic reference compounds.
Given the number and chemical diversity of organic extractables, it is unreasonable to expect that authentic reference compounds will be available (or can be made available) to confirm every identification. It is therefore necessary that levels of identification confidence be established and appropriately utilized. Data typically available from GC/MS and LC/MS analyses (see items a through e below) are used to designate individual extractables identifications in the categories of Confirmed, Confident, or Tentative (2, 5):
-
Mass spectrometric fragmentation behavior/expert mass spectrum interpretation
-
Confirmation of molecular weight
-
Confirmation of elemental composition
-
Mass spectrum matches automated library or literature spectrum
-
Mass spectrum and chromatographic retention index match authentic reference compound
-
Supporting spectral information from an orthogonal method (e.g., NMR)
-
A Tentative identification means that data have been obtained that are consistent with a class of molecule only. This is typically the case when only information such as a or d is available.
-
A Confident identification means that the tentative identification has been bolstered by additional and sufficient confirmatory information to preclude all but the most closely related structures. This would be the case, for example, if the tentative information (a and/or d) were augmented by b, c, or f. The more confirmatory information obtained, the greater the level of confidence.
-
A Confirmed identification means that the preponderance of evidence confirms that the entity in question can only be the identification that is provided. Although it is possible that a highly confident identification may meet the standard implied by the preponderance of evidence (for example, having a, b, c, e, and f), the only means of providing a confirmed identification is via mass spectral and retention time match with an authentic reference compound (item e).
-
A Tentative identification means that data have been obtained that are consistent with a class of molecule only. This is typically the case when only information such as a or d is available.
Although these identification categories are based on mass spectrometry, it is possible to use data from other analytical techniques to assist in extractables identification. Such techniques include GC/FTIR (Fourier Transform Infrared Spectroscopy) and LC/NMR (Nuclear Magnetic Resonance Spectroscopy). These and potentially other analytical techniques are capable of producing compound-specific data which are complementary to mass spectrometry.
The level of identification required for any individual extractable depends on the intended use of that identification. It is up to the organization responsible for the extraction study to determine this after appropriate consideration of applicable regulatory guidances.
Since the list of potential inorganic extractables, such as trace elements and metals, is finite compared with the population of organic extractables, identification and quantitative analysis for inorganic extractables are achieved simultaneously. While elemental analysis is relatively straightforward, it is not without its challenges. The issue of false positive responses and spectral or mass interference must be addressed in order for identifications based on atomic spectroscopy to be rigorous and accurate. It is also noted that elemental analysis provides element-specific identifications and quantitations, and not the chemical speciation of the extractable. Thus, interpretation of the impact of the elemental results may require further studies, such as detailed chemical speciation of elements deemed of significance. For example, while finding sulfur in an extract by atomic spectroscopy is an important outcome, the safety impact of this finding cannot be ascertained until the speciation of the sulfur is established. This is the case as the safety impact of sulfur as elemental sulfur may be different than that of sulfur as sulfate.
4. quantitation
Quantitation is typically based on the instrumental response of an individual extractable relative to an authentic reference compound, and therefore requires that individual extractables be separated (either directly with chromatography or indirectly with selective detection) and produce detector responses that are directly proportional to the level (or concentration) of the extractable in a given extract. Calibration of an analytical system is accomplished by analysis of authentic reference compounds (external standards). One or more internal standards can also be included in both the extract and reference calibration solutions to increase accuracy and precision. The levels of extractables for which authentic reference compounds are not available can be estimated using their responses (or response factors) relative to internal standards, or other surrogate reference compounds of similar molecular structure. While such an analytical process can provide reliably accurate concentration estimates, diligence must be exercised in terms of establishing and justifying the choice and use of internal standards. Criteria for the selection of appropriate internal standards have been described (2, 5).
Preparation of Extracts for Analysis
Extracts can often be analyzed directly without significant preparation or concentration. Many organic solvent extracts (e.g., dichloromethane, ethyl acetate, hexane) can be directly injected into a gas chromatograph, while others (e.g., methanol, ethanol, isopropanol) are either too reactive in the heated GC injection port or too high boiling. Organic solvent extracts with inappropriate physical/chemical properties for direct analysis by GC can be switched to more appropriate solvents. Certain extractables, such as fatty acids (e.g., palmitic acid, stearic acid) perform better in gas chromatographic analysis when they are derivatized to either methyl esters or trimethylsilyl esters.
It is usually considered inappropriate to directly analyze aqueous extracts by gas chromatography, due to the reactivity and high boiling point of water. In addition, pH-buffered aqueous extracts contain nonvolatile salts which are not suitable for GC injection. Aqueous extracts are typically back extracted with an organic solvent to remove organic extractables from the water, with the resulting organic extract being injected into the GC. Unlike GC, liquid chromatography (HPLC, LC/MS) is perfectly suited to the direct analysis of aqueous extracts, since most HPLC methods include water and water-miscible mobile phases. Water-immiscible organic solvents (e.g., hexane) cannot be injected onto these reversed-phase HPLC systems, so these must be dried and the resulting extractable residue taken up in a solvent suitable for HPLC (e.g., acetonitrile, methanol, or mixtures of these with water).
Organic or aqueous extracts with insufficient levels of extractables for analysis can be concentrated by various techniques. Many organic solvents can be dried down under inert gas, a rotary evaporator, or a Kuderna-Danish concentrator. Aqueous extracts can be lyophilized, concentrated under vacuum, or back extracted into an organic solvent which is then further concentrated.
The final concentration at which an extract is analyzed depends on the goal(s) of the extractables assessment and the inherent sensitivities of the analytical techniques applied. A good “rule of thumb” is that in order to accomplish a complete structural analysis of an unknown extractable, a GC/MS requires approximately 5 ng injected into the instrument. This suggests a concentration in the injected extract of 5 ng/µL or 5 µg/mL. In a 200-mL dichloromethane extract, this converts to a total of 1 mg of this particular extractable recovered from the extracted test article. If this analyte concentration is insufficient to meet the goal of the extractables assessment, then the following parameters can be optimized:
-
Extraction stoichiometry (i.e., extract more material or use more extracting solvent)
-
Extraction conditions (i.e., use higher temperatures, longer times, solvents with greater extraction power, more aggressive extraction technique, etc.)
-
Extract processing (i.e., concentration of the extract)
SUMMARY
Assessing the Completeness of an Extractables Assessment
The completeness of an extractables assessment can only be judged against the overall goals of the assessment. For example, an extractables assessment accomplished solely for materials characterization might include one extracting solvent, one extraction technique, and one set of extraction conditions; along with a materials-based threshold (e.g., 10 ppm w/w). Such an extractables assessment might be considered complete if all extractables above the defined threshold were identified to the confident level (defined above) and quantitated. For an extractables assessment designed to establish a rigorous leachables–extractables correlation for a high risk drug product, where a challenging safety threshold might apply (e.g., 0.15 µg/day; see reference 5), good scientific practice and due diligence requires the following:
-
Generation of extracts should be accomplished with
-
Multiple solvents or extracting media with varying extracting power based on the known extracting power of the drug product vehicle;
-
Multiple and complementary extraction techniques, including those with the capability for volatiles analysis;
-
Extraction conditions that allow equilibrium to be achieved.
-
Multiple solvents or extracting media with varying extracting power based on the known extracting power of the drug product vehicle;
-
Characterization of extracts should use
-
Multiple and complementary analytical techniques;
-
Careful sample preparation, keeping the analytical technique(s) in mind;
-
A systematic process for identification and quantitation of extractables.
-
Multiple and complementary analytical techniques;
In this case, the extractables assessment might be considered complete if all extractables above the defined threshold were identified to at least the confident level, quantitated, and correlated both qualitatively and quantitatively with drug product leachables data (if available) and the known ingredients in the packaging system, packaging component(s), or material(s) of construction.
It should be noted that limited extractables assessments with relatively narrow goals can be accomplished to required completeness with a relatively focused effort. For example, extraction studies designed to quantitate the levels of specific chemical additives in specific packaging components/materials can be done with specified extraction parameters and analytical methods (see Containers—Plastics 661). The reader is also referred to various sources which describe extractables assessments and extraction studies for pharmaceutical applications (2, 5, 6), as well as other general sources which refer to extractables assessments for medical devices (9) and food contact (11).
Reference is also made to compendial chapters in this Pharmacopeia which include extraction studies with specific goals and purposes:
Example Extractables Profiles—Materials Characterization
As stated previously, extraction studies are usually designed to produce extractables profiles, which are qualitative and/or quantitative analytical representations of the extractables content of a particular extracting medium. To illustrate the concept of an extractables profile, the following is an example of an extractables study performed for the purpose of material characterization. Extractables profiles are commonly produced by analysis of laboratory extracts by instrumental chromatographic techniques. Figure 2 and Figure 3 show example extractables profiles (GC/MS and HPLC/UV, respectively) from a hexane Soxhlet extract of a cyclic olefin copolymer (COC) material of construction (12). COC materials are used in the fabrication of pre-filled syringes, vials for small-volume parenterals, and bags for large-volume parenterals. The extracts were generated by subjecting approximately 5 g of suitably sized material to 16 hours of Soxhlet extraction with 125 mL of hexane. The resulting extract was spiked with internal standards (details not relevant to this discussion) and was analyzed directly by GC/MS (see Table 4). For HPLC/UV analysis, an aliquot of the hexane extract was reduced in volume and diluted with methanol prior to analysis (see Table 5). Since the purpose of this extractables assessment was materials characterization, extraction study parameters were adjusted relative to an extractables identification threshold of 10 ppm (µg/g). It is clear from these chromatograms that the techniques used are both complementary and orthogonal, illustrating the concept that multiple analytical methods are required to typically elucidate the complete extractable profile.
+'&img=../../images/v38332/c1663_fig2_pf395.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 2. GC/MS chromatogram (extractables profile) for a hexane Soxhlet extract of a cyclic olefin copolymer (12). Internal standards (IS) producing peaks in this chromatogram include: 2-Fluorobiphenyl at 10.7 min, Irganox 415 at 22.0 min, and Bisphenol M at 23.0 min. Numbered peaks represent identified extractables above the materials-based threshold.
+'&img=../../images/v38332/c1663_fig3_pf395.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 3. HPLC/UV Chromatograms (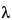 = 220 nm; extractables profile) for a hexane Soxhlet extract of a cyclic olefin copolymer (12). Internal standards (IS) producing peaks in this chromatogram include: Bisphenol M at 7.6 min, 2-Fluorobiphenyl at 7.8 min, and Irganox 415 at 8.7 min. The major peak in this extractables profile above the materials-based threshold is the known additive (antioxidant) Irganox 1010 (Peak 2).
= 220 nm; extractables profile) for a hexane Soxhlet extract of a cyclic olefin copolymer (12). Internal standards (IS) producing peaks in this chromatogram include: Bisphenol M at 7.6 min, 2-Fluorobiphenyl at 7.8 min, and Irganox 415 at 8.7 min. The major peak in this extractables profile above the materials-based threshold is the known additive (antioxidant) Irganox 1010 (Peak 2).
Table 4. Operating Parameters, GC/MS Analysis of the Hexane COC Soxhlet Extract (12)
| Operating Parameter |
Operating Value |
|---|---|
| Column |
J&W DB-5HT, 30-m × 0.25-mm, 0.25 µm film thickness |
| Oven Program |
Start at 50 |
| Carrier Gas |
He at 1.2 mL/min |
| Injection |
Split (1:5); 1 µL |
| Injector Temperature |
300 |
| FID Detector Temperature |
N/A |
| MS Transfer Line Temperature |
180 |
| MS Detection Details |
70 eV (+) EI (electron ionization), mass range of 33–650 amu (3.0-min solvent delay) |
Table 5. Operating Parameters, HPLC/UV Analysis of the Hexane COC Soxhlet Extract (12)
| Operating Parameter |
Operating Value |
|
|---|---|---|
| Column |
Agilent Zorbax Eclipse Plus C18, 100- × 3.0-mm, 3.5-µm particles |
|
| Column Temperature |
40 |
|
| Mobile Phase Components |
A = 10 mM ammonium acetate, B = acetonitrile |
|
| Mobile Phase Gradient |
Time |
%B |
| 0.0 |
5.0 |
|
| 8.4 |
100.0 |
|
| 35.0 |
100.0 |
|
| 36.0 |
5.0 |
|
| 39.0 |
5.0 |
|
| Mobile Phase Flow Rate |
0.8 mL/min |
|
| Sample Size |
10–50 µL |
|
| Detection, UV |
205–300 nm; spectra recorded at |
|
REFERENCES
-
FDA. Guidance for industry: container–closure systems for packaging human drugs and biologics. Rockville, MD: FDA; 1999.
-
Ball D., Norwood D., Stults C., Nagao L., eds. Leachables and Extractables Handbook. New York: J. Wiley and Sons; 2012.
-
ANSI/AAMI. BE 823:2006 Biological evaluation of medical devices—part 18: chemical characterization of materials. 2006. http://webstore.ansi.org/RecordDetail.aspx?sku=ANSI%2fAAMI+BE83%3a2006+(R2011)+(ANSI%2fAAMI+BE+83%3a2006+(R2011)). Accessed 19 March 2013.
-
European Medicines Agency. Guideline on plastic immediate packaging materials. 2005. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003448.pdf. Accessed 19 March 2013.
-
PQRI Leachables and Extractables Working Group. Safety thresholds and best practices for extractables and leachables in orally inhaled and nasal drug products. 2006. http://www.pqri.org/pdfs/LE_Recommendations_to_FDA_09-29-06.pdf. Accessed 19 March 2013.
-
Jenke D. Compatibility of Pharmaceutical Solutions and Contact Materials: Safety Considerations Associated with Extractables and Leachables. New York: John Wiley and Sons; 2009.
-
ANSI/AAMI/ISO. 10993-12:2012. Biological evaluation of medical devices—part 12: sample preparation and reference materials. 2012. http://marketplace.aami.org/eseries/scriptcontent/docs/Preview%20Files/10993-12_1207_preview.pdf. Accessed 19 March 2013.
-
Martin J., Fitzgerald R., Pothier N., Ding W. Recommendations for Testing and Evaluation of Extractables from Single-Use Process Equipment. Washington, DC: Bio-Process Systems Alliance; 2010.
-
ANSI/AAMI/ISO. 10993-12:2007. Biological evaluation of medical devices—part 12: sample preparation and reference materials. 2007. https://marketplace.aami.org/eseries/scriptcontent/docs/Preview%20Files%5C10993-12_preview.pdf. Accessed 19 March 2013.
-
ASTM International. ASTM F 1980-07 Standard guide for accelerated aging of sterile barrier systems for medical devices. 2011. http://webstore.ansi.org/RecordDetail.aspx?sku=ASTM%20F1980-07(2011)&source=msn&adgroup=astm. Accessed 19 March 2013.
-
21 CFR 177 Indirect Food Additives: Polymers.
-
Pennino S., Norwood D. Controlled extraction study on cyclic olefin copolymer (COC). 2011. http://www.pqri.org/workshops/PODP11/pdfs/posters/Cyclic_Olefin_Copolymer.pdf. Accessed 19 March 2013.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Desmond G. Hunt, Ph.D.
Senior Scientific Liaison (301) 816-8341 |
(GCPS2010) General Chapters - Packaging Storage and Distribution |
USP38–NF33 Supplement : No. 1 Page 7166
Pharmacopeial Forum: Volume No. 39(5)