



INTRODUCTION
This chapter describes best practices for production, conjugation, and characterization of polysaccharide and glycoconjugate vaccines. It describes key quality attributes at each step of the process and suggests best methods to assess these attributes. The scope of this chapter includes vaccines consisting of one or more purified polysaccharides (such as pneumococcal, meningococcal, and Typhoid Vi vaccines) and components involved in their production, and vaccines consisting of one or more glycoconjugate immunogens in which a saccharide has been covalently attached to a suitable carrier protein. The latter category includes Haemophilus influenzae type b (Hib), meningococcal, and pneumococcal conjugate vaccines. The chapter does not include combination vaccines in which Hib conjugates are combined with unrelated immunogens against diphtheria, tetanus, and pertussis.
BACKGROUND
Occurrence of Capsular Polysaccharides
Many pathogenic bacteria possess a polysaccharide capsule that encloses the cell, modulates the flow of nutrients to the cell surface, and protects against dehydration. When a bacterium establishes an infection in a mammalian host, the polysaccharide capsule hides cell surface components from elements of the mammalian immune system, such as antibodies and complement proteins that otherwise would activate mechanisms to kill the pathogen. Although polysaccharide capsules are themselves immunogenic in children and adults, development of a protective immune response may be too slow to defend the host against disease. In many cases, antibodies directed against the capsular polysaccharide are protective, and the prior existence of these antibodies prevents establishment of the infection; this is the basis for their use as vaccines or components of conjugate vaccines. Many bacterial species can be divided into serogroups or serotypes that express structurally and immunologically distinct capsular polysaccharides. The number of known serotypes differs between organisms. There are six known Haemophilus influenzae serotypes and more than 90 Streptococcus pneumoniae serotypes. Different serotypes (or serogroups) of the same organism may have different infectivities. For instance, the large majority of disease caused by H. influenzae is due to the b serotype. Different serotypes may cause disease in different geographical regions, may produce a different spectrum of disease, or may be prevalent in different age groups. The serotype-specific pattern of disease may change with time within a given geographical region. For these reasons, many polysaccharide and conjugate vaccines contain multiple saccharide immunogens.
Structures of Capsular Polysaccharides
Capsular polysaccharides are high molecular weight polymers that contain a strict repeat unit. This repeat unit can be a single monosaccharide unit or can be an oligosaccharide unit that contains as many as eight sugar residues. The repeat units can be linear or branched, and are sometimes linked together by phosphodiester bonds. Bacterial polysaccharides often contain unusual sugar residues that are not found elsewhere, and may be substituted with a wide range of acylating groups (O-acetyl groups are the most common) and phosphorylated substituents such as phosphoglycerol. Incomplete O-acetylation and migration of O-acetyl groups between different hydroxyl groups leads to a degree of heterogeneity in the polysaccharides.
Immune Responses to Capsular Polysaccharides
Capsular polysaccharides are T-cell–independent type 2 immunogens. These immunogens do not evoke antibody isotype switching, affinity maturation, or immunological memory. Because of the relatively late development of the relevant arm of the human immune system, unconjugated polysaccharide vaccines usually induce only a poor immune response in infants under the age of two and thus are not used for this population group. Adjuvants in polysaccharide vaccines do not improve the immune response. In a typical adult target population only a single dose is required to induce a protective immune response. In the absence of immunological memory, regular revaccination is required, often at five-year intervals. Repeated vaccination can lead to hyporesponsiveness to the vaccine.
Conjugation of Polysaccharides to Carrier Protein
Covalent attachment of a capsular polysaccharide, or an oligosaccharide derived from it, to a protein carrier creates a conjugate vaccine. Immunization with a conjugate vaccine induces humoral immunity by means of a different molecular mechanism that does not require cross-linking of immune cell surface proteins. For this reason, effective conjugate vaccines can be produced using oligosaccharide haptens that may be derived from a capsular polysaccharide or from inherently lower molecular weight polysaccharides such as the O-chain of a lipopolysaccharide. Conjugates can also be produced using naturally high-mass polysaccharides or by controlled size reduction to shorter chains. Depending on the manufacturing process there are three basic structural models for conjugate vaccines:
- conjugates in which a carrier protein is modified with multiple oligosaccharide chains that have one or two activation sites to allow attachment to a carrier protein, resulting in a monomeric glycoconjugate or a glycoconjugate with limited cross-linking
- cross-linked conjugates in which multiple activated polysaccharide chains and carrier proteins couple to multiple polysaccharide chains, creating a cross-linked network of proteins and glycans
- conjugates in which a size-reduced polysaccharide is covalently attached to a complex of proteins, typically bacterial outer proteins, via multiple attachments.
The Carrier Protein
The most widely used carrier proteins are related to bacterial toxins that are detoxified by chemical or genetic means. Conjugate vaccines induce a T-cell–dependent response that is developed early in life and leads to immunological memory and boosting of the response by further doses of the vaccine, thus they are suitable for infant immunization. The role of the carrier protein in modulating the immune response is discussed below.
Conjugation Chemistry
Polysaccharides can be covalently attached to proteins, although activation of the polysaccharide is required. Sometimes the carrier protein is also activated to create compatible reactive groups. Polysaccharides can be activated initially by creation of reactive aldehyde groups, by periodate oxidation or uncovering of the reducing terminal of sugars, by reaction of hydroxyl groups with highly reactive reagents such as cyanogen bromide (or 1-cyano-4-dimethylamino-pyridinium tetrafluoroborate [CDAP] as a crystalline alternative) or carbonyldiimidazole, by use of carboxylic acid groups, or, if available, by free amino groups or phosphate groups. Periodate oxidation, dilute acid hydrolysis, and some other approaches to polysaccharide activation may, depending on the structure of the polysaccharide, lead to depolymerization of oligosaccharides, which are typically fractionated by size and an appropriate fraction coupled to the carrier. The “natural” reactive groups on proteins include lysine  -amino groups, N-terminal amino groups, carboxylic acids in aspartate or glutamate, or thiol groups. Carrier protein activation may also involve the creation of hydrazide groups or thiols for linkage. Covalent attachment between the activated polysaccharide and the carrier protein, activated or not, can take place via the use of activated esters, sometimes created in situ with a water-soluble carbodiimide reagent, by reduction of Schiff's bases, by reaction of a thiol with maleimide, or by elimination of bromide. Conjugation can either be direct, as in reductive amination, or via the introduction of a suitable bifunctional linker. In general, the choice of conjugation chemistry is defined by either the structure of the polysaccharide repeat unit or the desire to produce a conjugate of a specific structural family.
-amino groups, N-terminal amino groups, carboxylic acids in aspartate or glutamate, or thiol groups. Carrier protein activation may also involve the creation of hydrazide groups or thiols for linkage. Covalent attachment between the activated polysaccharide and the carrier protein, activated or not, can take place via the use of activated esters, sometimes created in situ with a water-soluble carbodiimide reagent, by reduction of Schiff's bases, by reaction of a thiol with maleimide, or by elimination of bromide. Conjugation can either be direct, as in reductive amination, or via the introduction of a suitable bifunctional linker. In general, the choice of conjugation chemistry is defined by either the structure of the polysaccharide repeat unit or the desire to produce a conjugate of a specific structural family.
Immune Responses to Conjugate Vaccines
The immune response to the saccharide component of conjugate vaccines is T-cell–dependent and is similar to the response for proteins, although this process is still not fully understood with respect to glycoconjugates. Because this immunological pathway is in place even in infants, conjugate vaccines were developed initially for use in infants. Following interaction with antigen-presenting cells such as dendritic cells, macrophages, and B-cells, glycoconjugate vaccines are internalized and processed into small peptides and glycopeptides that then are re-exposed and presented to T-lymphocytes in association with the major histocompatibility complex class II molecules. Multiple immunizations of an immunologically naive infant are normally required to raise an antibody response, but processes such as isotype switching and affinity maturation take place and immunological memory is stimulated. In general terms, these processes result in induction of long-lasting high-affinity antibodies that are effective at preventing bacterial pathogens from establishing an infection. Adjuvants in glycoconjugate vaccines are effective at boosting immune responses. These vaccines have also been shown, in the cases of Hib, meningococci, and pneumococci, to eliminate nasopharyngeal carriage of organisms, thus an important aspect of their effectiveness arises from suppressing transmission of infectious serotypes between individuals, called a herd effect.
KEY QUALITY PARAMETERS FOR BULK POLYSACCHARIDES
Bulk monovalent polysaccharide is purified from bacterial cell culture and is a key stable intermediate in the manufacture of both polysaccharide and conjugate vaccines. Many quality parameters for final vaccines can be initially assessed by analysis of bulk monovalent polysaccharides or by using critical in-process tests supporting the manufacturing process. Important quality parameters for purified polysaccharide include identity, purity, composition, and molecular size, and depend on the type and extent of further processing. The purification process is validated to consistently produce compliant material.
The purity of the polysaccharides depends on the purification steps, including harvest methods, clarification, and downstream purification processes. For purification, a combination of precipitation, filtration, and chromatographic procedures can be used, depending on the chemical nature of the polysaccharide. The final purification step can consist of buffer exchange and filtration followed by storage of purified polysaccharide (frozen), or additional precipitation and washing of the precipitate with solvent before drying, followed by storage. Drying of polysaccharides can be performed in dessicators and can include several steps of grinding or fluffing and return to the dessicators for further drying. Manufacturers should take care during these steps because mechanical handling of the polysaccharide can reduce its molecular size. The purified polysaccharide is stored at a suitable temperature in conditions that avoid the uptake of moisture. Lyophilization of polysaccharides is also possible. The stability of the polysaccharide under specified storage conditions should be demonstrated; this may include assessment of the optimal moisture content of the dried material.
Differentiating which tests should be used for polysaccharide release and which are better suited to in-process testing to ensure process consistency depends on the process and how the polysaccharide will be further processed. Final decisions about process parameters typically take place after discussions with regulatory authorities.
Dry Weight
Because dry weight is used both to calculate the results of certain tests with reference to the dried substance and to calculate amounts for subsequent processes, volatile matter, including water, in the purified polysaccharide is determined by a combination of suitable methods including the following:
-
thermogravimetry (see Loss on Drying
 731
731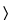 )
)
-
Karl Fischer (water only: see Water Determination 921)
-
residual solvents determined by gas chromatography (see Residual Solvents 467 and Chromatography 621) and by nuclear magnetic resonance (NMR) spectroscopy (see Nuclear Magnetic Resonance Spectroscopy 761) or colorimetric methods.
Polysaccharide Identity
The manufacturer is required to confirm the identity of the active component present in the purified polysaccharide. If other polysaccharides are produced at the same manufacturing site, the method should be validated to show that it distinguishes the desired polysaccharide from all other polysaccharides produced in that facility (see 21 CFR 610.14). The identity is determined by a prespecified combination of suitable methods such as:
-
Immunological test methods: These methods require access to highly specific antisera that are able to distinguish between closely related polysaccharide antigens. Commonly used formats for this purpose are immunoprecipitation, immunoelectrophoresis, and enzyme-linked immunosorbent assays (ELISA). The (serological) specificity of the antiserum should be demonstrated by the absence of cross-reactivity with heterologous polysaccharides manufactured in the same facility. More information regarding these methods can be found in Immunological Test Methods—General Considerations 1102, Immunological Test Methods—Enzyme–Linked Immunosorbent Assay (ELISA) 1103, and Immunological Test Methods—Immunoblot Analysis 1104.
-
NMR spectroscopy (see 761): NMR methods require access either to authentic samples of the polysaccharide or to reference spectra. Comparison is made visually in terms of the positions, relative intensities, and multiplicities of significant resonances, or by use of other objective methods.
- Polysaccharide identity can also be assessed by use of a matrix of compositional assays, usually with colorimetric or chromatographic readouts, that define factors such as the amounts of different sugar types (e.g., amino sugars, uronic acids, or methyl pentoses depending on the polysaccharide), the amount of substituents (e.g., O-acetyl groups), and the content of nitrogen and phosphorus.
Polysaccharide Purity and Quantity
colorimetric assays
Historically, polysaccharide content compared to dry weight (purity) was estimated using a range of colorimetric assays for functional groups specific to the polysaccharide antigen. Because the reference standard is typically a pure monosaccharide or similar model compound, and because assays were typically determined before most structures were known, component quantification may not parallel true stoichiometry. Thus, manufacturers should develop correlations among alternative approaches. Purity can be calculated based on the method employed and the salt form present. Table 1 lists colorimetric tests that may be appropriate to determine the composition of a particular polysaccharide within a vaccine. The response factors for the sugar units in the polysaccharide may differ from those of a pure monosaccharide reference standard. Manufacturers should address these concerns during method validation.
Table 1. Colorimetric Assays for Polysaccharide Composition and Quantity
| Orcinol Assay (Bial Reagent) | Molybdate (Chen) Assay | Resorcinol Assay (Seliwanoff Reagent) | Sulfuric Acid Digestion | Carbazole Assay | Dimethyl- aminobenz- aldehyde Assay |
Cysteine Sulfuric Acid Assay | Hestrin Assaya |
Anthrone-Sulfuric Acid Assay | |
|---|---|---|---|---|---|---|---|---|---|
| PS Antigen | Ribose | Phosphate | Sialic Acid | Total Nitrogen | Uronic Acids | Hexosamines | Methylpentoses | O-Acetyl | Total Sugar |
| Hib PRP | X | X | |||||||
| MenA | X | X | |||||||
| MenC | X | X | |||||||
| MenY | X | X | X | ||||||
| MenW135 | X | X | X | ||||||
| Pneumo (Serotype Specific) | X | X | X | X | X | X | X | X | |
| Vi | X | ||||||||
|
a
The hestrin assay is appropriate for compositional analysis and identity, but because the degree of O-acetylation can vary between polysaccharide batches it is not normally a suitable assay for polysaccharide quantitation.
|
|||||||||
hydrolysis and composition analysis by chromatography
Acid or base hydrolysis depolymerizes polysaccharides into oligosaccharides, monosaccharides, or smaller fragments that are polysaccharide-specific for the optimized hydrolysis conditions employed. Aggressive hydrolysis conditions can destroy some components of the polysaccharide. These fragments can be quantified directly by use of, for example, high-performance anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD) or conductivity detection for ions (HPAEC-CD), or by reversed phase high-performance liquid chromatography (HPLC) of fluorophore-labeled sugars. Alternatively, the hydrolysate can be derivatized and analyzed by gas chromatography with flame ionization (GC-FID) or mass spectrometric detection (GC-MS). Suitable polysaccharide reference materials or monosaccharides are required, and the hydrolysis conditions are product-specific. Table 2 summarizes the type of hydrolysis applied, the analytical methods, and the analytes that have been quantified for different polysaccharides.
Table 2. Chromatographic Methods for Compositional Analysis and Quantification of Vaccine Polysaccharidesa
| PS Antigen | Acid Hydrolysis and HPAEC |
Acid Hydrolysis, Fluorophore Labeling, and HPLC |
HF Hydrolysis and HPAEC |
Base Hydrolysis and HPAEC |
Methanolysis and GC or HPAEC |
|---|---|---|---|---|---|
| PRP | Ribitol | — | Phosphate | PRP monomer | — |
| MenA | ManN-6-P | — | Phosphate | O-acetyl | — |
| MenC | Neu5Ac | — | — | O-acetyl | — |
| MenY | Glc, Neu5Ac | Neu5Ac | — | O-acetyl | — |
| MenW135 | Gal, Neu5Ac | Neu5Ac | — | O-acetyl | — |
| Pneumo (Serotype Specific) |
Alditols, methylpentoses, hexoses, hexosamines, uronic acids, Pyruvate | — | Phosphate | O-acetyl | Alditols, methylpentoses, hexoses, hexosamines, uronic acids |
| Vi | — | — | — | O-acetyl Vi monomer fragment | — |
|
a
PS = polysaccharide; HF = hydrofluoric acid; PRP = polyribosylribitol phosphate; MenA = Meningococcus group A; MenC = Meningococcus group C; MenY = Meningococcus group Y; MenW135 = Meningococcus group W135; Pneumo = Pneumococcus; Man = mannose; Neu = neuraminic acid; Ac = acetyl; Glc = glucosamine; Gal = galactose.
|
|||||
capillary electrophoresis
Capillary zone electrophoresis has been used for the identification and quantification of meningococcal polysaccharides without depolymerization.
immunochemical assays
Immunochemical assays such as immunonephelometry or ELISA require access to specific antisera that must be calibrated to reference materials. The response may be modified by the matrix or size of the polysaccharide.
nmr spectroscopy
The relative intensities of characteristic resonances can confirm the proportions of different sugar residue types and substituents such as N- or O-acetyl or pyruvate that are present in the polysaccharide. Quantification can be achieved by comparison of these intensities to that of an added internal standard (see 761).
Regarding phosphorus determination, one should remember that a number of bacterial polysaccharides contain phosphodiester linkages. The polysaccharide may be quantified based on its phosphorus content by colorimetric assays or instrumental approaches such as inductively coupled plasma–optical emission (ICP-OES) or ICP-mass spectrometry (ICP-MS).
counterions
If the percentage purity of polysaccharide bulks (compared to dry weight) is part of their release program, the amount and type of counterion present must be considered and can be determined by, for example, ICP-MS. This is normally an in-process control step.
Polysaccharide Molecular Size Distribution
The molecular size distribution is generally evaluated by liquid chromatography using soft gel–filtration procedures or size exclusion–high-performance liquid chromatography (SEC-HPLC) equipped with in-line refractive index (RI). The results are reported as the distribution coefficient (KD) determined from the main peak of the elution curve or as the percentage of material that elutes before a defined KD cut-off value. The absolute molecular weight of the polysaccharide and its hydrodynamic (gyration) radius can be determined by coupling static light scattering and RI detectors to the SEC-HPLC column, and measuring the RI increment (dn/dc) using reference polysaccharides. Requirements based on molecular weight can be expressed in terms similar to those based on molecular size, and related either to peak values or to the proportion that elutes before a defined cut-off value.
Level of Protein Contamination
The residual protein content of the polysaccharide should be determined by an appropriate assay and should be shown to be below the approved specification. These specifications typically vary by polysaccharide and serotype. Method validation should assess the need for sample pretreatment before protein determination and specific interference by the polysaccharide in the protein assay. Further, method validation should demonstrate that the assay sensitivity is appropriate for the specification. Biotechnology-Derived Articles—Total Protein Assay 1057 contains information regarding these assays. Methods that are typically applied to polysaccharides include colorimetric assays (e.g., Lowry, Biuret, bicinchonic acid, or Bradford assays) and UV absorbance.
Level of Nucleic Acid Contamination
The nucleic acid content of the purified polysaccharide should be determined and should be shown to meet specifications, which are typically less than 1% w/w. Nucleic Acid–Based Techniques—Approaches for Detecting Trace Nucleic Acids (Residual DNA Testing) 1130 provides additional information about these methods.
Quantification of Process- and Product-Related Impurities
Depending on in-process testing or release requirements, chromatographic and spectroscopic methods can be used to quantify residuals from the fermentation and isolation/purification steps. These residuals may include antifoaming agents, phenol, cetyltrimethyl ammonium bromide, ethanol, and other residual solvents. Other impurities should also be quantified using appropriate tests, including:
-
bacterial endotoxins (see Bacterial Endotoxins Test 85)
-
pyrogens (see Pyrogen Test 151)
-
sterility (see Sterility Tests 71), if required
-
bioburden (see Microbiological Examination of Nonsterile Products: Microbial Enumeration Tests 61), where appropriate.
As an in-process control, the proportion of pneumococcal C polysaccharide in pneumococcal capsular polysaccharides can be determined by a combination of 1H and 31P NMR spectroscopy or by HPAEC-PAD analysis of ribitol. Alternatively, it can be derived from the compositional analysis.
Stability-Indicating Tests
The polysaccharide may lose integrity because of gradual hydrolysis, resulting in a reduced molecular size, and this degradation can be monitored by size exclusion chromatography or by high-performance size exclusion chromatography (HPSEC) with static light scattering detectors for molecular weight. Loss of O-acetyl groups can be monitored by HPLC methods. Both the loss or migration of O-acetyl groups and the integrity of phosphate-containing polysaccharides can be tracked by NMR spectroscopy. Immunochemical methods can also be used to monitor polysaccharide integrity but should be validated for that purpose.
KEY QUALITY PARAMETERS FOR BULK FORMULATED POLYSACCHARIDE VACCINE
The bulk formulated polysaccharide vaccine refers to a solution that contains a blend of appropriate amounts of the required monovalent bulk polysaccharides, as well as any buffer salts, excipients, adjuvants, and antimicrobial preservatives present in the vaccine product. Typically, the vaccine is sterilized, usually by filtration, and is ready for filling in the final dosage forms.
Antimicrobial Preservative
If an antimicrobial preservative is present and is not assayed at a later stage, its concentration can be assayed at this stage using an appropriate validated chemical, physicochemical, chromatographic, or spectroscopic assay. While the specification will depend on the regulatory agency, typically the amount of preservative should not exceed 120% of the expected value, and the manufacturer must demonstrate that the preservative is effective at that concentration (see Antimicrobial Effectiveness Testing 51).
Sterility
At this stage, if the expectation is that the material is sterile, then sterility should be demonstrated using an appropriate validated assay such as one of those described in 71. For other manufacturing processes, measurement of some combination of bioburden, endotoxin count, or pyrogenicity may be sufficient.
Polysaccharide Quantity
To ensure correct dilution of the bulk before final filling, and unless other control mechanisms are in place, determination of the content of individual serotypes or total polysaccharide content may be required using appropriate validated physicochemical or immunochemical methods as defined in the bulk polysaccharide section.
pH, Osmolarity/Isotonicity, and Excipients
The final format of a vaccine may be liquid or lyophilized. If the format in this stage does not change during the final fill and is not analyzed later then manufacturers should assay this bulk material for:
-
pH (see pH 791)
-
possibly osmolarity or isotonicity (see Osmolality and Osmolarity 785). If approved by a regulatory agency, routine osmolarity testing may be omitted if the manufacturer demonstrated consistency in development and clinical lots.
- excipient content, if present.
KEY QUALITY PARAMETERS FOR POLYSACCHARIDE VACCINES: FINAL FILLS
In accordance with 21 CFR 610.1, “No lot of any licensed product shall be released by the manufacturer prior to the completion of tests for conformity with standards applicable to such product. Each applicable test shall be made on each lot after completion of all processes of manufacture which may affect compliance with the standard to which the test applies. The results of all tests performed shall be considered in determining whether or not the test results meet the test objective, except that a test result may be disregarded when it is established that the test is invalid due to causes unrelated to the product.” Test methods should be appropriately verified and validated.
Description and Solubility
Liquid vaccines are typically clear colorless liquids that are essentially free from visible particles. Lyophilized products are typically white or cream-colored powders or pellets that are freely soluble in water and yield clear colorless liquids that are free from visible particles.
Polysaccharide Identity
The manufacturer should demonstrate that all the expected polysaccharides are present in the final fill. The test may be immunochemical, physicochemical, or chemical. Regulation 21 CFR 610.14 requires that the identity test should distinguish the product from other products handled in the same facility. In some cases specific quality attributes of the polysaccharide relating to identity, such as O-acetyl content, may also be specified, and should be assayed at this final fill stage if not assessed at an earlier stage. Often polysaccharide identity and quantity can be confirmed and determined by the same assay.
Polysaccharide Quantity
The content of each polysaccharide present in the final lot should be determined by a suitable validated immunochemical or physicochemical method. Typically, Vi and pneumococcal polysaccharide vaccines contain 25 µg of each serotype in a single human dose, whereas meningococcal polysaccharide vaccines contain 50 µg of each serogroup in a single human dose. When immunochemical methods are used, the antisera should be specific for each polysaccharide in the vaccine, including, in the case of the pneumococcal serotypes, immunologically cross-reactive species. Specifications are established on a case-by-case basis, but typically the content of each polysaccharide in the vaccine should be either between 70% and 130% or between 80% and 120% of the label claim.
Polysaccharide Structural Integrity and Molecular Size
In the absence of suitable validation data showing that no changes occur during filling and storage, the structural integrity and molecular size of the individual polysaccharide components should be assessed as far as is possible following the final fill. Depending on the nature of the polysaccharide and the complexity of the vaccine, the integrity of individual serotypes or serogroups can be established by a combination of immunochemical stability-indicating measurements (e.g., ELISA, rate nephelometry, or physicochemical methods), by size or molecular weight determination alone or in combination with serotype-specific assays, or by quantification of specific groups such as O-acetyl groups that have been shown to be critical for immunogenicity. Together with the serotype-specific quantification, these assays act as a surrogate for a potency assay.
pH
The pH of the final fill should be determined for liquid products or for redissolved lyophilized products according to 791 and should meet the requirements of the relevant licensing authority. This attribute should be included in stability-testing programs.
Antimicrobial Preservative
Where applicable, the amount of antimicrobial preservative should be determined by a suitable validated approach. Typically, the value should not exceed 120% of the quantity stated on the label. The approved lower limit should not be lower than the minimum amount shown to be effective throughout the product's shelf life.
Process Impurities
Unless the product has been tested at an earlier manufacturing stage, and depending on the manufacturing process used, process impurities or residuals such as phenol or formaldehyde should be tested by appropriate validated assays. For more information about allowable process impurities, see Vaccines for Human Use—Bacterial Vaccines 1238.
Sterility
The sterility of each lot should be ensured according to procedures described in 71 and 21 CFR 610.12. The product should comply with the requirements of the tests.
Osmolarity and/or Isotonicity, Excipients, and Moisture Content
If the vaccine is a liquid preparation, the pH and osmolarity/isotonicity of each final lot should be tested and shown to be within the pre-approved specifications. For a lyophilized preparation, analysts should measure the pH after reconstitution with the appropriate diluent.
Excipient functional category (sometimes referred to as functionality) is a broad, qualitative, and descriptive term for the purpose or role an excipient serves in a formulation. Of greater importance, however, are the quantitative performance requirements (i.e., critical material attributes) of excipients that must be evaluated and controlled to ensure consistent performance throughout the product life cycle. Manufacturers should anticipate lot-to-lot and supplier-to-supplier variability in excipient properties and should have in place appropriate controls if needed to ensure consistent excipient performance (refer to Excipient Performance 1059 for guidance).
Diluent for Lyophilized Products
Data should be provided to support diluent sterility (see 71) and to ensure that adventitious microbial contamination does not occur under the reconstitution conditions (i.e., diluents should not introduce contamination) or during storage conditions as described in the package insert. If an antimicrobial preservative is used (as is the case normally only in multidose products), testing according to 51 is recommended to demonstrate acceptability. Testing may not be required on all lots once process control and consistency have been established.
General Safety
Depending on regulatory requirements, a general safety test may be necessary as set out in 21 CFR 610.11(g), and the product should meet the specifications.
KEY QUALITY PARAMETERS FOR CARRIER PROTEIN
A number of protein carriers have been used in preclinical and clinical evaluation of conjugate vaccines. Proteins such as diphtheria and tetanus toxoids, which derive from the respective toxins after chemical detoxification with formaldehyde, initially were selected as carriers because of the safety track record accumulated with tetanus and diphtheria vaccination, and these proteins are used today as carriers for meningococcal, Hib, and pneumococcal vaccines in a number of countries worldwide. CRM197, a nontoxic mutant of diphtheria toxin, is also used as a carrier for licensed Hib, pneumococcal, and meningococcal conjugate vaccines and for other vaccines being developed. An outer membrane protein complex (OMPC) of serogroup B meningococcus is the carrier for a licensed Hib conjugate vaccine. An Hib-related protein, Protein D, is the carrier for most of the polysaccharides included in one licensed conjugated pneumococcal vaccine.
Key Quality Parameters for Carrier Protein
Five carrier proteins currently are used for conjugate vaccines approved for use by various regulatory authorities: Diphtheria Toxoid, Tetanus Toxoid, CRM197, Haemophilus Protein D, and OMPC. When the carrier protein is a component of an approved vaccine like diphtheria and tetanus toxoids, the first key quality parameters are those defined by the release tests on the concentrated bulks for these components. Other quality parameters include the level of oligomerization (monomer vs. multimeric forms). For carrier proteins that are not licensed as stand-alone vaccines, the list of key quality attributes should at least include identity, sterility or bioburden (depending on the manufacturing process), endotoxins, and purity. In some specific cases, additional quality attributes may require measurement.
carrier protein identity
The identity of the carrier proteins can be assessed by suitable methods that can be divided into two categories:
- Immunochemical methods: immunoprecipitation (flocculation, radial immunodiffusion, and nephelometry), immunoelectrophoretic methods (rocket immunoelectrophoresis), and immunoenzymatic methods (immunoblots and ELISA)
- Standard physicochemical methods used for other purified proteins: mass spectrometry, peptide mapping, and molecular mass determination.
Using these methods, a sample preparation is compared to a reference preparation to demonstrate consistency. The tests listed here may not be appropriate for toxoid proteins.
endotoxins
To ensure an acceptable level of endotoxin in the final product, manufacturers can determine the endotoxin content of the carrier protein according to 85 and thus can show that endotoxin levels are within acceptable limits. For some products, rabbit pyrogenicity testing (151) may be a more relevant test.
Diphtheria Toxoid
antigenic purity
Depending on the manufacturing process diphtheria toxoid preparation can show different degrees of purity. Typically, antigenic purity for diphtheria toxoid as determined by the flocculation test should be at least 1500 Lf (limit of flocculation) units/mg of protein.
monomer, dimer, or aggregate content
Diphtheria toxin is characterized by the presence of dimeric and multimeric aggregation forms that are also present in the corresponding detoxified preparations. Regarding this parameter, analysts can monitor the manufacturing consistency of diphtheria toxoid by determining the content of monomers vs. dimers and other aggregates using a suitable method such as SEC-HPLC coupled with a static light scattering detector. In some cases, because of the low purity of the preparation the HPLC profile may result in a broad peak that cannot be resolved into the contribution of the individual species.
Tetanus Toxoid
antigenic purity
Typically, the antigenic purity of tetanus toxoid as determined by the flocculation test should be at least 1500 Lf units/mg of protein.
monomer, dimer, or aggregate content
The detoxification process for tetanus toxin results in oligomerization to an extent that depends on the process conditions. Similar to diphtheria toxoid, the physicochemical consistency of tetanus toxoid can be monitored by the determination of monomeric vs. dimeric forms and other aggregates using suitable methods like SEC-HPLC coupled with static light scattering detection. Other methods like ultracentrifuge analysis can be applied but may be less suitable for routine testing.
CRM197
CRM197 protein is a nontoxigenic diphtheria toxin isolated from the supernatant of cultures of Corynebacterium diphtheriae C7(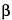 197)tox- and purified by a sequence of chromatographic and diafiltration steps. A guideline for production and control of bacterial proteins used in vaccine manufacturing is given in 1238. Recombinant CRM197 should meet requirements for nonrecombinant material, although additional characterization appropriate for recombinant proteins may be required.
197)tox- and purified by a sequence of chromatographic and diafiltration steps. A guideline for production and control of bacterial proteins used in vaccine manufacturing is given in 1238. Recombinant CRM197 should meet requirements for nonrecombinant material, although additional characterization appropriate for recombinant proteins may be required.
purity
The purity of CRM197 batches should be determined via suitable methods, e.g., HPLC (see 621), sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE; see Biotechnology-Derived Articles—Polyacrylamide Gel Electrophoresis 1056), or Capillary Electrophoresis 1053. Typical expectations are that the purity of CRM197 should be at least 90% and often greater than 95%.
degree of nicking
CRM197 contains an exposed loop of three arginine residues that is clipped by proteases present in the culture medium, resulting in a so-called nicked form. The manufacturing process should demonstrably be able to regularly produce CRM197 with a consistently low degree of nicking. In the presence of a reducing agent like dithiothreitol, the nicked form breaks down into two distinct polypeptides called fragments A and B that can be easily detected by SDS-PAGE, which accordingly is a suitable method to determine the degree of nicking. In a validated process testing may be needed as an in-process control.
Haemophilus Protein D
Haemophilus Protein D is obtained as a recombinant protein from E. coli fermentation that, after extraction from the cells, is purified by a series of chromatographic and diafiltration steps and finally is sterile filtered. Routine release tests for protein D include identity, purity, sterility, protein content, and endotoxin content. Host cell proteins and host cell DNA should also be tested unless process validation has shown consistent clearance.
purity
Purity should be monitored with an appropriate test such as HPLC, SDS-PAGE, or capillary electrophoresis (CE).
Outer Membrane Protein Complex
The OMPC of Neisseria meningitidis group B is derived from meningococcal serogroup B bacterial cells by extraction with buffer-containing detergent. Analysts can monitor manufacturing consistency by determining the OMPC composition with SDS-PAGE or another suitable method.
endotoxin content or pyrogenicity
To ensure an acceptable level of endotoxin in the final product, analysts can determine the endotoxin content of the carrier protein and can show that it falls within acceptable limits according to 85. Alternatively, OMPC preparations should pass the rabbit pyrogenicity test following injection into rabbits at, typically, 0.25 µg/kg of body mass (see 151).
meningococcal ompc requirements—purity and lipopolysaccharide content
The composition of meningococcal OMPC carrier should be monitored for consistency by SDS-PAGE or by another suitable method. Typically, the lipopolysaccharide (LPS) content should not exceed 8% by weight. Suitable methods for LPS determination include HPLC, colorimetric analyses, SDS-PAGE, and GC-MS.
KEY QUALITY PARAMETERS FOR ACTIVATED INTERMEDIATES
Different chemical strategies can be applied to the synthesis of glycoconjugate vaccines. Two main approaches have traditionally been employed for glycoconjugate vaccines preparation: one is based on random chemical activation along the chain of the native or slightly size-reduced polysaccharide, followed by conjugation; the other is based on selective activation of end groups of oligosaccharides generated by controlled fragmentation of the native polysaccharide and subsequently coupling to the protein carrier. Depending on the conjugation chemistry, a chemical spacer can be used to facilitate the coupling of the protein to the saccharide antigens, and, in some cases, prior derivatization of the protein carriers is also required. In some cases the activated or derivatized polysaccharide or oligosaccharide is isolated and represents an intermediate of the glycoconjugate vaccine manufacturing process. In order to ensure a reproducible product by means of consistent application of the appropriate conjugation stoichiometry, manufacturers should use appropriate methods to determine the degree of activation or derivatization of the poly- or oligosaccharide and of the carrier protein.
Degree of Activation of Activated Polysaccharide
If appropriate, different activation or derivatization strategies can be applied to poly- or oligosaccharides in order to make them suitable for covalent coupling to the carrier protein. In some cases the hydroxyl groups of the polysaccharide first are reacted with cyanogen bromide or CDAP or carbonyldiimidazole to form active esters. These active intermediates then can be reacted in situ with adipic acid dihydrazide or other bifunctional amines to introduce an amino linker. Some bacterial polysaccharides possess carboxyl or phosphate groups that might be used for introduction of an amino linker using a carbodiimide-mediated chemistry. Subsequent coupling to the carboxyl groups of the carrier protein to obtain the desired glycoconjugate can be performed using, for example, a soluble carbodiimide-mediated chemistry. Alternatively, the amino linker incorporated into the polysaccharide structure can be further derivatized to obtain a bromo-acyl or a maleimido function that is amenable to coupling with a thiol group that is present or previously was incorporated into the carrier protein.
In some other cases aldehyde groups can be introduced into the polysaccharide structure by reaction of vicinal hydroxyl groups with the oxidizing reagent sodium metaperiodate (NaIO4). Depending on the polysaccharide structure, the NaIO4 treatment can also be used for simultaneous controlled depolymerization and aldehyde group generation. The poly- or oligosaccharides-containing aldehyde groups then can be covalently coupled to lysine residues and the N-terminal amino groups of the carrier protein by reductive amination in the presence of sodium cyanoborohydride (NaBH3CN) or other reducing agents that are selective for Schiff bases.
Some manufacturing strategies are based on the controlled hydrolysis of the native polysaccharides to produce oligosaccharides that can be specifically derivatized by a sequence of steps that lead to the introduction of an active ester function at their reducing ends. The desired conjugate is then obtained by reaction of the activated oligosaccharides with the lysine residues and N-terminal amino groups of the carrier protein.
To ensure a consistent conjugation stoichiometry, and therefore a consistent manufacturing process, manufacturers should determine the level of derivatization or activation of the poly- or oligosaccharide intermediates. Appropriate methods for determination of the newly introduced chemical functions into the saccharide structures should be in place and could include, for example, colorimetric or other suitable methods. In cases where the activated polysaccharide is conjugated without isolation, consistency in the degree of polysaccharide activation may also be demonstrated as part of process validation or reflected by characteristics of the final conjugate bulk.
The calculation of the degree of poly- or oligosaccharide derivatization may require determination of the total saccharide quantity that can be achieved by applying, for example, HPAEC-PAD, colorimetric assays, or other suitable methods. In a validated process where production consistency has been established, and depending on the conjugation chemistry used and the results of clinical trials, testing may be used as an in-process control. Residual unconjugated linker that could interfere with subsequent steps should be controlled via measurement or process validation.
Molecular Sizing of Activated, Derivatized, or Processed Polysaccharide
The molecular size and degree of polymerization of the poly- or oligosaccharide intermediates depends on the particular manufacturing process and should be measured because these attributes can affect the consistency of the conjugation process. Suitable tests should be applied to intermediate pools that are selected on the basis of the different manufacturing processes. Examples of suitable methods for profiling molecular sizes and determining the degree of polymerization of poly- and oligosaccharides are: SEC coupled with UV, RI, or static light scattering detectors; colorimetric assays based on total and end group determination; HPAEC-PAD; or NMR spectroscopy. In a validated process, testing can be used as an in-process control.
Degree of Activation of Activated Carrier Protein
As mentioned above, some manufacturing procedures for glycoconjugate vaccines may also require activation of the protein carrier. This process step introduces into the protein side chains additional functional groups that react with the poly- or oligosaccharide intermediates activated with the proper functional group. In general, such functional groups are introduced by derivatization of protein amino acid side chains like glutamic or aspartic acid with a bifunctional reagent (e.g., adipic acid dihydrazide or hydrazine) so that a highly nucleophilic hydrazide group becomes available for coupling with the polysaccharide. In other manufacturing strategies, the lysine side chains of the carrier protein can be derivatized to introduce different reactive groups (e.g., bromo-acyl, thiol, or maleimido groups). Appropriate methods for determination of the newly formed chemical functions introduced into the carrier proteins should be in place and may include spectrophotometric assays and mass spectrometry. The calculation of the degree of activation or derivatization of the carrier protein may also require the determination of the total protein quantity (e.g., by colorimetric assays or other appropriate methods). In a validated process where production consistency has been established, and depending on the conjugation chemistry used and the results of clinical trials, testing may be used as an in-process control. Depending on the conjugation chemistry used (i.e., immediate conjugation after activation), consistency in degree of carrier protein activation may also be demonstrated as part of process validation or reflected by characteristics of the final conjugate bulk.
Carrier Protein Monomer Content
In some cases the procedures for protein carrier activation or derivatization may result in a certain degree of covalent aggregation of the carrier itself, and this should be monitored with appropriate tests like SEC-HPLC coupled with static light scattering detection, SDS-PAGE, matrix-assisted laser desorption–ionization mass spectrometry, or other suitable tests.
KEY QUALITY PARAMETERS FOR MONOVALENT BULK CONJUGATE (DRUG SUBSTANCE)
Conjugation
Conjugation of the polysaccharide antigen to the carrier protein or protein complex is the critical component of the manufacturing process for conjugate vaccines. A general overview of a conjugation process is presented in 1238. In addition to developing a description of critical processing equipment, reagents, and processing steps, manufacturers should provide the rationale for the conjugation chemistry selected and the purification steps, if any, used to remove unwanted reaction components. Clearance of product-related impurities (e.g., unconjugated polysaccharide or unconjugated carrier protein) should be monitored and controlled.
Depending on the nature of the manufacturing process, the monovalent conjugate bulks can be considered to be the drug substance or can be considered a process intermediate.
During conjugation, the reactive functional groups present on the polysaccharide antigen are reacted with the functional groups located on the carrier protein or carrier complex to form stable, covalent bonds. Many different types of chemistries are used, including reductive amination, thio-alkylation, or CDAP chemistry. The choice of chemical method should be based on the availability of functional groups, either naturally occurring or introduced via an activation or side chain loading process, and the ability to control the manufacturing process to produce a consistent and stable product.
Although the conjugation process may be conceptually straightforward, the process should be well controlled. The process typically consists of mixing the activated polysaccharide with the selected carrier and allowing the components to react. Depending on the nature of the chemical reaction, an additional chemical reactant may be needed to complete the reaction or stabilize the conjugate product. For example, in the case of reductive amination, it may be necessary to add a reducing agent to convert the linkage from a relatively unstable Schiff base to a more stable secondary amine. Chemical deactivation or capping of residual reactive groups may also be required. Finally, residual levels of unreacted components such as free protein, free polysaccharide, chemical reagents, and byproducts should be removed from the process via validation or should be monitored by testing. Regulatory authorities may request stability evaluation of these intermediates because the data may support stability predictions for multivalent vaccines for which data collection is more difficult.
To define and control the conjugation process, the manufacturer should establish targets for process parameters and tolerances for all critical process steps where possible, including extent of activation, charge ratios for each reaction component, reaction time, reaction temperature, reaction pH, and mixing conditions. Additionally, tolerances for the purity of each of the reaction components should be established, including the polysaccharide, carrier protein, and any chemical components as noted above.
The common key quality attributes for monovalent conjugates, the rationale for monitoring these parameters, and suitable test methods are described below.
Polysaccharide Identity
Polysaccharide identity confirms that the correct antigen was used during the manufacturing process and that no critical epitope was lost during conjugation. Polysaccharide identity should be confirmed using a suitable immunological or chemical method. Examples of immunological methods include ELISA, immunoblot analysis, and rate nephelometry. The specificity of the test method must be ensured by selection of appropriate reagents. Alternatively, the identity of the polysaccharide can be confirmed using a chemical or physical method such as HPLC, HPAEC-PAD, GC, or NMR if acceptable specificity can be demonstrated and it can be shown that the carrier protein does not substantially interfere with the identification of the polysaccharide.
Carrier Protein Identity
Depending on the nature of the manufacturing process and the manufacturing controls, it may be necessary to confirm the identity of the carrier protein, e.g., during a manufacturing process for a multivalent product in which different antigens are conjugated to different carrier proteins within the same facility. Carrier protein identification can also be performed using an immunological method such as ELISA or, if possible, an appropriate chemical method such as peptide mapping (see Biotechnology-Derived Articles—Peptide Mapping 1055). If appropriate, the carrier identity can be evaluated in the same assay that is used for the identity of polysaccharide.
Polysaccharide Quantity
Polysaccharide quantity or concentration must be confirmed for all lots of monovalent conjugate because it is directly related to the product dose. Polysaccharide yield can also be a useful marker for process consistency. A variety of methods are available and suitable for use in determining the polysaccharide concentration. These include colorimetric methods such as the phenol–sulfuric acid, orcinol, and anthrone–sulfuric acid assays and monosaccharide analysis following hydrolysis by HPAEC-PAD, HPLC with fluorescence detection (HPLC-FD), or GC. Immunological methods that may be suitable include ELISA or rate nephelometry. The suitability of these methods depends on the availability of appropriate reagents. The choice of method should be made on the basis of precision and accuracy. Interference from the carrier protein must be avoided. Additionally, the chemistry of the polysaccharide antigen should be considered when analysts select the method. For example, the phenol–sulfuric acid assay may not be suitable for use if the antigen is composed largely of amino sugars or sialic acid. Suitable methods for polysaccharide quantification are listed in previous sections of this chapter.
Carrier Protein Quantity
The concentration of the carrier protein must be confirmed for all lots of monovalent conjugate. Conjugate vaccines are typically formulated based on the polysaccharide concentration, not the carrier protein concentration. However, the concentration of the carrier protein is needed to determine the polysaccharide–protein ratio, a key indicator of process consistency.
Analysts should select a test method that is specific for the carrier protein and does not suffer from interference from the polysaccharide components. Suitable methods may include amino acid analysis (see Biotechnology-Derived Articles—Amino Acid Analysis 1052), colorimetric protein tests such as the bicinchoninic acid assay or UV absorbance (see 1057 for both types of methods), or the protein-specific output from HPSEC with static light scattering, RI, or UV detection.
Polysaccharide–Protein Ratio
As noted in Carrier Protein Quantity, the polysaccharide–protein ratio may be an indicator of process consistency. Therefore, tolerances should be established for two of the three parameters: polysaccharide concentration, protein concentration, and polysaccharide–protein ratio. It is not necessary to establish limits for all three parameters because the polysaccharide–protein ratio is typically calculated from the measured polysaccharide and carrier protein concentrations. Because the downstream processing and dilutions usually are based on the amount of polysaccharide present in the monovalent bulks, the limit for the polysaccharide content can be based on the minimum concentration required for downstream processing.
Molecular Size Distribution or Integrity or Proof of Covalency
The molecular size of the conjugate is a key indicator of process consistency. Unusually small conjugates may indicate incomplete conjugation whereas unusually large conjugates may indicate aggregation and may result in a loss of yield during downstream filtration steps. The average molecular size and the size distribution should be measured using appropriate sizing methods such as SEC, HPSEC, HPSEC with static light scattering or RI detection, or analytical ultracentrifugation. The choice of the method should be made on the basis of the expected size of the conjugate and the availability of a chromophore for detection.
Proportion of Free (or Unconjugated) Polysaccharide
The proportion of unconjugated polysaccharide must be monitored for each lot of monovalent conjugate because of the possibility that the presence of a large amount of unconjugated polysaccharide may suppress the immune response to the antigen. Additionally, the presence of free polysaccharide is a key indicator of process consistency and is an indirect measure of covalent attachment to the carrier. Measurement of the proportion of unconjugated polysaccharide can be used as a stability-indicating test if appropriately validated.
To measure the level of unconjugated polysaccharide, analysts must separate the unconjugated polysaccharide from the conjugated polysaccharide. This can be achieved chromatographically, or by chemical precipitation of the conjugate with acid or detergents, aluminum adsorption, capillary electrophoresis, gel filtration, centrifugal ultrafiltration, solid-phase extraction, or immunoprecipitation. The amount of free polysaccharide then can be quantitated using the method that was used to quantitate the total polysaccharide level, if that method is sufficiently sensitive, by UV detection if a chromophore is present, or by immunological or appropriate physicochemical methods.
The level of unconjugated polysaccharide must be measured at release and during stability testing because deconjugation is a potential degradation mechanism.
Proportion of Unconjugated Carrier Protein
The level of unconjugated carrier protein must be monitored for each lot of monovalent conjugate because this level is a key marker of process consistency and is an indirect measure of covalency. To measure the amount of unconjugated protein, the unconjugated protein must be separated from the conjugate. This can be done chromatographically or by electrophoresis (slab or capillary). Once it is separated, the amount of unconjugated protein can be monitored by UV or by a colorimetric method (see 1057). Method selection should be based on sensitivity, precision, and specificity for unconjugated protein.
Unreacted Functional Groups
During the conjugation process, reactive functional groups on the polysaccharide react with functional groups on the carrier protein. However, the reaction is typically not driven to completion, and a process of capping of remaining reactive groups may be required, depending on the nature of the residual reactive groups, the conjugation chemistry used, and manufacturing process optimization. The method chosen to cap reactive groups depends on the conjugation chemistry employed. Even so, some reactive groups may still remain on the conjugate even after the reaction is quenched by reduction, or after remaining reactive groups have been chemically capped. Safety concerns, if any, depend on the nature of the reactive groups and the level of reactive groups that remain. The level of residual reactive groups should be monitored as a measure of process consistency unless process validation has shown that unreacted functional groups detectable at this stage are removed during subsequent manufacturing processes. Additionally, the presence of residual reactive groups may affect product stability during storage.
The test method used to evaluate residual reactive groups depends on the activation chemistry that is used and the nature of the polysaccharide antigen. Appropriate methods may include gas chromatography, HPLC with fluorescence, or UV detection following hydrolysis.
Residual Reagents
Consistency in the amount of residual reagents from the conjugation chemistry can be demonstrated during process development, and the process can be validated for their clearance. This validation includes not only unconjugated polysaccharide and protein but also buffers, salts, small-molecule reaction components, and byproducts generated during conjugation. Provided that consistent levels of residual solvents are recovered, such testing may serve as an in-process control.
Sterility or Bioburden
Depending on the manufacturing process, the monovalent conjugate bulks should be tested for bioburden or sterility. If the monovalent conjugate bulks are subjected to additional process steps with no process holds, in some cases it may be appropriate to perform the sterility or bioburden test at a downstream step.
Bacterial Endotoxins
The monovalent conjugate bulks must be tested for bacterial endotoxins. If the monovalent conjugate bulks are subjected to additional process steps with no process holds, in some cases it may be appropriate to perform the endotoxin test at a downstream step.
FORMULATED AND ADJUVANTED (IF APPROPRIATE) CONJUGATE BULKS
Monovalent conjugate bulks can be individually adsorbed and formulated as monovalent bulks before mixing during preparation of the final vaccine.
Adjuvant Content
If an adjuvant has been added to the conjugate bulk, its content should be determined by an appropriate method. If aluminum is used as an adjuvant, typical maximum values are 0.85 mg of aluminum per dose, although higher limits up to 1.25 mg of aluminum per dose may be accepted if justified, and lower limits may apply according to governing agency requirements.
Polysaccharide Content
Assessment of polysaccharide content can be required but is often difficult at this stage because of the presence of adjuvants or excipients. Several methods are available and are suitable for use in determining the polysaccharide concentration. These include colorimetric methods such as the phenol–sulfuric acid, orcinol, and anthrone–sulfuric acid assays; and post-hydrolysis monosaccharide analysis by HPAEC-PAD, HPLC-FD, or GC. Suitable immunological methods include ELISA or rate nephelometry. If possible, the same method must be used for both this step and the conjugate bulk step. The choice of method should be based on precision, accuracy, and specificity. Interference from the matrix must be avoided. Additionally, the chemistry of the polysaccharide antigen should also be considered when analysts select the method.
Free (or Unconjugated) Polysaccharide
Free polysaccharide content must be measured as a release test for monovalent formulated bulk if the free polysaccharide cannot be accurately or precisely measured in the final product. For most applications, polysaccharide testing is a marker of consistency; i.e., it is monitored for each bulk in order to establish production consistency. The test may be omitted when manufacturing consistency has been demonstrated, or the test can be used as an in-process control. If free polysaccharide could be adsorbed on adjuvant, a desorption step using, for example, phosphate buffer, may be required. The unconjugated polysaccharide must be separated from adsorbed and nonadsorbed conjugated polysaccharide. Adsorbed conjugate can be removed by centrifugation, and nonadsorbed conjugate can be eliminated chromatographically; by chemical precipitation of the conjugate with acid or detergent precipitation or aluminum adsorption; by immunochemical precipitation with anti–carrier antibodies; by capillary electrophoresis; or by gel or membrane filtration or ultrafiltration. The amount of free polysaccharide then can be quantitated using the same method that was used to quantitate the total polysaccharide level if the latter is sufficiently sensitive and accurate. Quantitative tests for polysaccharide include colorimetric methods such as the phenol–sulfuric acid, orcinol, and anthrone–sulfuric acid assays; and monosaccharide analysis following hydrolysis by HPAEC-PAD, HPLC-FD, or GC. Suitable immunological methods include ELISA or rate nephelometry.
Results should be expressed as the percentage of unconjugated polysaccharide vs. the total content experimentally determined, or, if this value cannot be determined experimentally, it may be possible to calculate it from a theoretical value.
The level of unconjugated polysaccharide must be measured during stability studies because deconjugation is a potential degradation mechanism.
Level of Adsorption to Adjuvant
The level of adsorption in monovalent formulated bulks must be performed as a release test if it cannot be performed on the final product. If it is not a release test then the adsorption test is a marker of consistency, and the test is performed on each bulk in order to establish production consistency. After consistent production has been demonstrated, the test can be omitted.
After product centrifugation, nonadsorbed conjugate is quantified in the supernatant. The adsorbed conjugate then is quantified by a suitable validated method, which can be the same test used for the total conjugate content. If the physical or chemical method used for the quantification is not specific for the conjugated form of the saccharide, the amount of free polysaccharide is subtracted from the total polysaccharide in order to determine the quantity of adsorbed conjugate. Otherwise an immunologically specific method can be used (e.g., ELISA). Results can be expressed as the percentage of nonadsorbed conjugate vs. the total content experimentally determined. If it is not possible to determine the amount of nonadsorbed conjugate experimentally then it may be possible to calculate it from a theoretical value. The level of adsorption to adjuvant must be measured during stability testing.
Sterility
The sterility of each lot should be measured according to procedures described in 71 and 21 CFR 610.12.
KEY QUALITY PARAMETERS FOR CONJUGATE VACCINE DRUG PRODUCT
Description and Solubility
Each container in each final fill or drug product should be inspected visually (manually or with automatic inspection systems), and containers that show abnormalities such as improper sealing, lack of integrity, or turbidity should be discarded. Similarly, the presence of clumping or particles may indicate a product failure.
Polysaccharide Identity
Polysaccharide identity tests confirm that the correct antigen was used during the manufacturing process. Polysaccharide identity should be confirmed using a suitable immunological or physicochemical method, e.g., ELISA, immunoblots, or rate nephelometry. The specificity of the test method must be ensured by use of appropriate reagents. Acceptable specificity must be demonstrated, and tests must show that the carrier protein does not substantially interfere with the identification of the polysaccharide. Assays based on hydrolysis and chromatographic identification of saccharide components (e.g., HPAEC) after polysaccharide hydrolysis may be acceptable.
Polysaccharide Quantity
Typically, monovalent conjugate vaccines contain 10 µg of saccharide, and multivalent vaccines contain between 1 and 10 µg of each serotype or serogroup per single human dose. Assessment of the content of polysaccharide may be difficult because of the presence of adjuvant or excipients, especially when multiple components are present. The amount of each polysaccharide may be required in order to calculate the free polysaccharide content and the proportion of unadsorbed conjugate, or this information may be used during processing of the vaccine's final formulation. Polysaccharide quantity or concentration must be confirmed for all types of monovalent conjugates because it is directly related to the product dose. A variety of methods are available and suitable for use in determining the polysaccharide concentration [see discussion of polysaccharide quantitation in the Key Quality Parameters for Monovalent Bulk Conjugate (Drug Substance) section above].
Carrier Protein Identity (if Appropriate)
Depending on the nature of the manufacturing process and the manufacturing controls, if the identity of the carrier protein has not been confirmed at an earlier stage, it may be necessary to do so before product release. For example, an identity test for the carrier protein could be necessary during the manufacturing process for a multivalent product for which different antigens are conjugated to different carrier proteins within the same facility. Carrier protein can be identified using an immunological method such as an immunoblot or ELISA, or using an appropriate chemical method such as peptide mapping. If appropriate, the carrier's identity can be evaluated in the same assay used to identify the polysaccharide.
Molecular Size (if Feasible or Appropriate)
If the molecular size distribution has not been established for the individual monovalent bulk conjugates used in the drug product formulation, the molecular size distribution of the conjugates must be determined in the final fill or drug product. The molecular size of the conjugate is a key indicator of process consistency. The average molecular size and the size distribution should be measured using appropriate sizing methods such as SEC, HPSEC, HPSEC with static light scattering or RI detection, analytical ultracentrifugation, or dynamic light scattering. The choice of the method should be based on the expected size of the conjugate and the availability of a chromophore or fluorophore for detection. In the case of molecular size distribution determinations at the final fill or in drug products composed of multivalent polysaccharides, serotype-specific detection methods may be required for the individual monovalent conjugates. Molecular size is a sensitive indicator of conjugate stability, and, where possible, it should be measured during stability studies.
Proportion of Free (or Unconjugated) Polysaccharide
To measure the level of unconjugated polysaccharide, analysts must separate the unconjugated polysaccharide from the adsorbed and nonadsorbed conjugated polysaccharide and interfering substances. If unconjugated polysaccharide may be adsorbed to adjuvant, prior desorption with, for example, phosphate buffer is required. Methods for separation and measurement of conjugated and unconjugated polysaccharide are described above in Formulated and Adjuvanted Conjugate Bulks. The level of unconjugated polysaccharide is a stability-indicating measurement because deconjugation is a potential degradation mechanism and should be measured during stability studies. However, assessing the level of unconjugated polysaccharide and the stability of complex multivalent products is technically demanding, and alternative approaches to assessing antigen integrity include molecular size, O-acetyl content, or immunological measurement. Specific assays may provide partial but overlapping information and should be matched to the product. Free saccharide data obtained for individual monovalent conjugates may also prove valuable and predictive.
pH, Osmolarity/Isotonicity, and Excipients
If the vaccine is a liquid preparation, the pH and osmolarity/isotonicity of each final lot should be tested and shown to be within the pre-approved specifications. For a lyophilized preparation, the pH should be measured after reconstitution with the appropriate diluent. Residual moisture in lyophilized products should be determined. See comments above regarding excipient functionality.
Adjuvant Quantities (if Appropriate)
If an adjuvant has been added to the conjugate bulk, its content should be determined by an appropriate method. If aluminum compounds (such as aluminum hydroxide or hydrated aluminum phosphate) are used as adjuvants, the amount of aluminum should not exceed 1.25 mg per single human dose or as otherwise required by the governing agency (see also 21 CFR 610.15).
Antimicrobial Preservative (if Appropriate)
During product development manufacturers should consider the stability of the chosen preservative and possible interactions between the vaccine components and the preservative. If a preservative has been added to the vaccine, the content of preservative should be determined by an appropriate method (see Antimicrobial Agents—Content 341). The amount of preservative in the vaccine dose should be shown neither to have any deleterious effect on the antigen nor to impair the safety of the product in humans. If present, the amount must be NLT the minimum amount shown to be effective and typically should be NMT 120% of the amount stated on the label.
Moisture Content (Lyophilized Products)
If the vaccine is freeze-dried, the average moisture content should be determined by an appropriate method. Values should be within limits established during the product's stability studies. Typically, the average residual moisture content should be NMT 2.5%, and no vial should be found to have a residual moisture content of 3% or greater.
Sterility
The sterility of each lot should be determined according to procedures described in 71 and 21 CFR 610.12.
Diluent for Reconstitution of Lyophilized Vaccines
Manufacturers should generate data that show that adventitious microbial contamination does not grow under the reconstitution conditions (e.g., with diluents that will be used for reconstitution) or under specified storage conditions. A preservative is not normally required for single-dose vials when the product will be used soon after reconstitution, but multidose vials do require preservatives. Testing in alignment with 51 is recommended to demonstrate acceptability, but testing may not be required routinely after process control and consistency have been established.
General Safety or Abnormal Toxicity
The general safety or abnormal toxicity for vaccines should be established by appropriate evaluation and should be consistent with levels found to be acceptable in vaccine lots that were used in clinical trials.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Maura C Kibbey, Ph.D.
Senior Scientific Liaison, Biologics & Biotechnology (301) 230-6309 |
(GCBA2010) General Chapters - Biological Analysis |
USP38–NF33 Page 1518
Pharmacopeial Forum: Volume No. 39(4)