Add the following:
INTRODUCTION
Topically applied drug products fall into two general categories: those applied to achieve local action and those applied to achieve systemic effects after absorption through the skin into the blood circulation. Local action can occur at or on the surface of the application site (e.g., stratum corneum, ocular epithelium), in the underlying tissues (e.g., epidermis and/or dermis) and on subcutaneous tissues (e.g., muscle or joint).
Topically applied drug products include, but are not restricted to creams, gels, ointments, pastes, suspensions, lotions, foams, sprays, aerosols, solutions, and transdermal delivery systems (TDS, also known as patches). The definitions and descriptions of these dosage forms, and brief information on their composition and/or manufacturing process can be found in Pharmaceutical Dosage Forms  1151
1151 .
.
Procedures and acceptable criteria for testing topically applied drug products can be divided into those that assess general product quality attributes and those that assess product performance. The product quality attributes include the following: description, identification, assay (strength), impurities, physicochemical properties, uniformity of dosage units, water content, pH, apparent viscosity, microbial limits, antimicrobial preservative content, antioxidant content, sterility, if applicable, and other tests that may be product specific. Product performance testing assesses drug release and other attributes that affect drug release from the finished dosage form.
Although most topically applied drug products are semisolids, liquids, or suspensions, TDS are physical devices that are applied to the skin and vary in their composition and method of fabrication. TDS release their active ingredients by different mechanisms. They can be passive or active. This chapter covers only the tests related to passive TDS.
PRODUCT QUALITY TESTS FOR TOPICALLY APPLIED DRUG PRODUCTS
Universal Tests
Universal tests (see ICH Guidance Q6A—Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances, available at www.ich.org) are listed below and are applicable to all topically applied drug products.
Description:
A qualitative description of the drug product should be provided. The acceptance criteria should include the final acceptable appearance of the finished dosage form and packaging. A visual examination should identify changes in color, adhesive migration (i.e., cold flow) for TDS, separations, crystallization, etc., that are specific to the drug product. The description should specify the content or the label claim of the article. This is not a compendial test but is part of the manufacturer's specification for the drug product.
Identification:
Identification tests are discussed in General Notices and Requirements, 5.40. Identification tests should establish the identity of the drug or drugs present in the article and should discriminate between compounds of closely related structures that are likely to be present. Identity tests should be specific for the drug substance(s) (e.g., infrared spectroscopy). Near infrared (NIR) or Raman spectrophotometric methods also could be acceptable for the identification of the drug product (see Near-Infrared Spectrophotometry 1119 and Raman Spectroscopy 1120). Identification solely by a single chromatographic retention time is not specific.
Assay:
A specific and stability-indicating test should be used to determine the strength (content) of the drug product. In cases when the use of a nonspecific assay (e.g., Titrimetry 541) is justified, other supporting analytical procedures should be used to achieve overall specificity.
Impurities:
Process impurities, synthetic by-products, impurities associated with the adhesive (e.g., residual monomers), residual solvents (see Residual Solvents 467), heavy metals (see Heavy Metals 231), and other inorganic and organic impurities may be present in the drug substance and excipients used in the manufacture of the drug product and should be assessed and controlled. Impurities arising from the degradation of the drug substance and those arising during the manufacturing process of the drug product should also be assessed and controlled.
Specific Tests
In addition to the universal tests listed above, the following specific tests should be considered on a case-by-case basis.
Uniformity of Dosage Units:
This test is applicable for TDS and for dosage forms packaged in single-unit containers (see Uniformity of Dosage Units 905).
Water Content:
A test for water content should be included when appropriate (see Water Determination 921). This test is generally formulation dependent. Therefore, it is not included in the compendial drug product monograph but is part of the manufacturer's specification for the drug product.
Microbial Limits:
Microbial examination of nonsterile drug products is performed according to the methods given in general chapters Microbiological Examination of Nonsterile Products: Microbial Enumeration Tests 61 and Microbiological Examination of Nonsterile Products: Tests for Specified Microorganisms 62, unless the formulation itself is demonstrated to have antimicrobial properties. Acceptance criteria for nonsterile pharmaceutical products based on total aerobic microbial count (TAMC) and total combined yeasts and molds count (TYMC) are given in Microbiological Examination of Nonsterile Products: Acceptance Criteria for Pharmaceutical Preparations and Substances for Pharmaceutical Use 1111.
Antimicrobial Preservative Content:
Acceptance criteria for antimicrobial preservative content in multidose products should be established. They should be based on levels of antimicrobial preservative necessary to maintain the product's microbiological quality at all stages throughout its proposed usage and shelf life (see Antimicrobial Effectiveness Testing 51).
Antioxidant Content:
If antioxidants are present in the drug product, tests of their content should be established unless oxidative degradation can be detected by another test method such as impurity testing. Acceptance criteria for antioxidant content should be established. They should be based on the levels of antioxidant necessary to maintain the product's stability at all stages throughout its proposed usage and shelf life.
Sterility:
Depending on the use of the dosage form (e.g. ophthalmic preparations, products that will be applied to open wounds or burned areas), sterility of the product should be demonstrated as appropriate (see Sterilty Tests 71).
pH:
When applicable, topically applied drug products should be tested for pH at the time of batch release and at designated stability time points for batch-to-batch monitoring. Because some topically applied drug products contain very limited quantities of water or aqueous phase, pH measurements may not always be warranted.
This test is generally formulation dependent. Therefore, it is not included in the compendial drug product monograph but is part of the manufacturer's specification for the drug product.
Particle Size:
The particle size of the active drug substance(s) in topically applied drug products is usually determined and controlled at the formulation development stage. However, topically applied drug products should be examined for evidence of particle size alteration (i.e., appearance of particles, changes in particle form, size, shape, habit, or aggregation) of the active drug substance that may occur during the course of product processing and storage. Such examinations should be conducted at the time of batch release and at designated stability test time points for batch-to-batch monitoring because changes that are visually (macro- and microscopically) observable would likely compromise the integrity and/or performance of the drug product. These types of testing are generally formulation dependent. Therefore, such tests are not included in compendial monographs but are part of the manufacturer specification for the drug product.
SPECIFIC TESTS FOR OPHTHALMIC DRUG PRODUCTS
Ophthalmic dosage forms must meet the requirements of Sterility Tests 71. If the specific ingredients used in the formulation do not lend themselves to routine sterilization techniques, ingredients that meet the sterility requirements described under Sterility Tests 71, along with aseptic manufacture, may be used. Multiple-use ophthalmic preparations must contain a suitable substance or mixture of substances to prevent growth of, or to destroy, microorganisms accidentally introduced during the use of the product (see Added Substances under Ophthalmic Ointments 771), unless otherwise directed in the individual monograph or unless the formula itself is bacteriostatic and/or the delivery system promotes bacteriostasis. The finished ophthalmic preparation must be free from large particles and must meet the requirements for Leakage and for Metal Particles under Ophthalmic Ointments 771. The immediate containers for ophthalmic preparations shall be sterile at the time of filling and closing. It is mandatory that the immediate containers for ophthalmic preparations be sealed and tamper-proof so that sterility is ensured at the time of first use.
SPECIFIC TESTS FOR TOPICALLY APPLIED SEMISOLID DRUG PRODUCTS
Apparent Viscosity
Viscosity is a measure of a formulation’s resistance to flow and is an assessment of the rheological properties of the dosage form (e.g., semisolid dosage form). Because only Newtonian fluids possess a measurable viscosity that is independent of shear rate, semisolid pharmaceutical dosage forms which are non-Newtonian products exhibit an apparent viscosity.
The apparent viscosity of semisolid drug products should be tested at the time of batch release and initially at designated stability test time-points to set specifications for batch-to-batch and shelf life monitoring. Measurement procedures should be developed as outlined in Viscosity 911. For semisolids that show thixotropy and/or irreversible changes in viscosity after shearing, specific attention should be given to sample preparation procedures to minimize variability in the measurement of apparent viscosity caused by variable shear histories (e.g., mixing speed and temperature, filling operation, sample handling). Furthermore, for some products, it may be warranted to have apparent viscosity specifications at more than one set of conditions (e.g., bulk in-process stage, final packaged product, high and low shear rates, different temperatures).
Apparent viscosity specifications based on data obtained during product development and shelf life testing should be established for batch release and throughout their proposed shelf life.
The apparent viscosity test is formulation and/or process dependent. Therefore, it is not included in compendial drug product monographs but is part of the manufacturer’s specification for the drug product. Furthermore, the specifications for apparent viscosity of semisolid dosage forms at batch release and during stability testing may be different. Although the apparent viscosity of the finished drug product at the time of batch release must conform to the product development specifications, for stability testing, the apparent viscosity specifications for the drug product should be based on statistical assessment of the product over its shelf life.
Uniformity in Containers
Topically applied semisolid drug products may show physical separation during manufacturing processes and during their shelf life. To ensure the integrity of the drug product, it is essential to evaluate the uniformity of the finished product at the time of batch release and throughout its assigned shelf life.
products packaged in tubes
Within-tube content uniformity can be assessed in the following manner.
Carefully remove or cut off the bottom tube seal and make a vertical cut from the bottom to the top of the tube. Carefully cut the tube around the upper rim, open the two flaps and lay the flaps open to expose the product.
Inspect the product visually for the presence of phase separation, change in physical appearance and texture, and other properties described in the product test for Description. If there is no observable phase separation or change in physical appearance and texture, and if the product meets the Description acceptance criteria, proceed as described below. If the product exhibits phase separation and/or change in physical appearance or texture, the product fails the tube content uniformity test.
The procedures describe below can be modified depending on the sensitivity of the quantitative procedure used to assay the drug substance(s) present in the formulation.
For Multiple-Dose Products That Contain 5 g or More:
Procedure 1—
- Using a single tube, after visually inspecting the product remove an appropriate amount of product from the top, middle, and bottom portions of the tube. The sample size should be sufficient for at least one assay determination of the active ingredient(s). Carry out the assay test for the active ingredient(s) in each portion of the product, and evaluate the test results using Acceptance Criteria A.
- If the product fails Acceptance Criteria A, test 3 additional tubes from the same batch following step 1 described above, and evaluate all 12 test results using Acceptance Criteria B.
Procedure 2—
- Using two tubes, after visually inspecting the product, remove an appropriate amount of product from the top, middle, and bottom portions of each tube. The sample size should be sufficient for at least one assay determination of the active ingredient(s). Carry out the assay test for the active ingredient(s) in each portion of the tube, and evaluate the test results using Acceptance Criteria A.
- If the product fails Acceptance Criteria A, test 2 additional tubes from the same batch following step 1 described above, and evaluate all 12 test results using Acceptance Criteria B.
For Multiple-Dose Products That Contain Less Than 5 g of Product:
- Test the top and bottom portions of 2 tubes using Procedure 1 or Procedure 2 as described above. Evaluate the test results using Acceptance Criteria A.
- If the product fails Acceptance Criteria A, test 2 additional tubes from the same batch following step 1 described above, and evaluate all 8 test results using Acceptance Criteria B.
Tube (Container) Content Uniformity Test Acceptance Criteria:
In determining the relative standard deviation (RSD) from multiple tubes, first determine the variance from the three measurements for each tube and average across the tubes. The RSD is calculated using this average variance.
Acceptance Criteria A—
All assay results are within the range of 90%–110% of the product label claim and the RSD is NMT 6% or as specified in the product specification or in the compendial monograph. If the RSD is greater than 6%, use Acceptance Criteria B.
Acceptance Criteria B—
No assay result is outside the range of 90%–110% of the product label claim and the RSD of the 12 assay results is NMT 6% or as specified in the product specification or in the compendial monograph.
products packaged in containers other than tubes
For semisolid products packaged in a container other than a tube when the sampling method presented above cannot be used, other sampling methods are acceptable, such as the one described below for a jar.
- Select a suitable syringe of sufficient length to extend to the bottom of the container.
-
Remove and set aside the syringe plunger and cut off the bottom of the syringe barrel. Sampling should take place from a location to the left/right of the mid-line of the jar surface to preserve an undisturbed region on the other side for any additional investigation (See Figure 1).
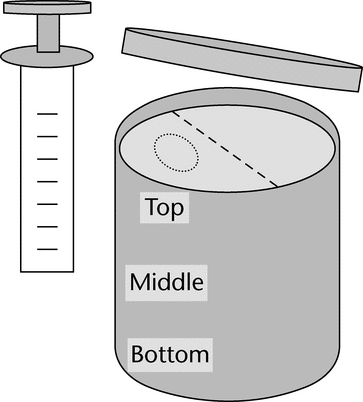 Figure 1. Sampling from a jar container.
Figure 1. Sampling from a jar container. - Slowly push the syringe barrel into the container until it reaches the bottom. Then, twist the syringe barrel containing the sample core, and remove the syringe from the container.
- Insert the syringe plunger into the barrel and carefully extrude the sample core onto a clean surface in three equal portions to represent the top, middle, and bottom portions of the container.
- Remove an appropriate sample representative of the middle section of the top, middle, and bottom portions of the container samples, and test according to the instructions outlined in Products Packaged in Tubes.
+'&img=../../images/v35300/m4033-pf363.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
SPECIFIC TESTS FOR TRANSDERMAL DELIVERY SYSTEMS
TDS or patches are formulated with an adhesive layer to ensure intimate contact with the skin to allow the delivery of the desired dose of drug. Adhesives in TDS must permit easy removal of the release liner before use, must adhere properly to human skin upon application, must maintain adhesion to the skin during the prescribed period of use, and must permit easy removal of the TDS at the end of use without leaving a residue or causing damage to the skin or other undesirable effect(s). Additionally, adhesives must be able to maintain the performance of the TDS throughout the shelf life of the drug product.
Three types of TDS adhesion tests are generally used: peel adhesion test (from a standard substrate), release liner peel test, and tack test.
Acceptance criteria are product specific and defined to assure that adhesion of each batch of TDS are within the range defined by the product design and are consistent between batches based on the product development specifications or statistical assessment of multiple product batches over the product's shelf life.
Peel Adhesion Test
This test measures the force required to remove (peel away) a TDS attached to a standard substrate surface (e.g., polished stainless steel). The TDS is applied to the substrate using specified techniques for application and is conditioned at a specified temperature and time. Then, the TDS is peeled away from the substrate with an instrument that allows control of peel angle (e.g., 90 or 180 degrees) and peel rate (e.g., 300 mm/min), and the peel force is recorded. This procedure is repeated using a minimum of five independent samples. The product fails the test if the mean peel force is outside the acceptable range determined during product development and/or based on statistical assessment of multiple product batches over the product's shelf life.
Release Liner Peel Test
This test measures the force required to separate the release liner from the adhesive layer of the TDS. The test is performed with a finished product sample. The test sample is conditioned using specific procedures (temperature and time). Then the release liner is pulled away from the TDS with an instrument that allows for control of peel angle (e.g., 90 or 180 degrees) and peel rate, and the peel force is recorded. This procedure is repeated using a minimum of five independent samples. The product fails the test if the mean peel force is outside the acceptable range determined during product development and/or based on statistical assessment of multiple product batches over the product's shelf life.
Tack Test
Several methods of tack tests have been developed. Examples include the probe tack method and the rolling ball method. It is up to the TDS manufacturer to decide which one is more appropriate for each drug product.
probe tack method
This test measures the force required to separate the tip of the test probe from the adhesive layer of the TDS. This test uses an instrument designed to create a bond between the tip of the stainless steel test probe of defined geometry and the TDS using a controlled force (light pressure) and specified test conditions (i.e., rate, contact time, contact pressure, temperature). Then, while controlling the rate of probe removal, the test measures the profile of force required to separate the probe tip from the TDS and the maximum force required to break the bond (tack). This procedure is repeated using a minimum of five independent samples. The product fails the test if the mean test result [force profile(s) and/or tack] is outside the acceptable range determined during product development and/or based on statistical assessment of multiple product batches over the product's shelf life.
rolling ball method
This test measures the distance travelled by a defined ball on the adhesive layer of the TDS under defined conditions, as a parameter dependent on the tack properties of the adhesive layer. This test uses a setup designed to roll a ball (with defined material, weight, size, and surface) from a ramp (with defined angle and length) onto the adhesive layer (with defined orientation) under specified test conditions (temperature) (see ASTM D3121 for more details). The distance traveled by the ball on the adhesive layer is measured using a suitable measuring device. This procedure is repeated using a minimum of five independent samples. The product fails the test if the mean distance travelled is outside the acceptable range determined during product development and/or based on statistical assessment of multiple product batches over the product’s shelf life.
Leak Test
This test is applicable only to form-fill-seal (reservoir or pouched)-type TDS. Form-fill-seal TDS must be manufactured with zero tolerance for leaks because of their potential for dose dumping if leaking occurs.
In-process control methods to examine TDS for leakers or potential leakers are needed and require considerable development on the part of TDS manufacturers.
in-process testing
During the manufacturing process, the presence of leakage, or potential for leakage, because of TDS perforation, cuts, and faulty seals resulting from failures such as air bubbles, gel splash, or misalignment of a TDS's backing and release liner layers, must be examined. Unless automated process analytical technology is implemented, in-process testing to identify these defects should be performed using the following test procedures:
Visual Inspection:
- A specified number of TDS, defined based on the batch size, should be randomly examined.
- Each sampled TDS should be thoroughly visually inspected for leakage.
- The product fails if any of the TDS examined is detected with a leak.
Seal Integrity:
Transdermal system seals should be stress tested to ensure that the application of pressure does not force seals to open, thereby leading to leakage.
- A specified number of TDS, defined based on the batch size, should be randomly examined.
- Each sampled TDS should be thoroughly visually inspected for leakage.
- Each sampled TDS is placed on a hard, flat surface and overlaid with a weight so that it is subjected to 13.6 kg. The weight should be left in place for 2 minutes. Upon removal of the weight, the TDS should be visually inspected for leakage.
- The product fails if the number of TDS detected with a leak is greater than the acceptable limit established by the manufacturer.
Packaged Product Testing:
TDS may leak after they have been individually placed in the primary packaging material as a result of the packaging operation itself or by user opening of the packaging. Therefore, TDS should be tested for leakage after they have been manufactured and packaged in their primary packaging material.
- A specified number of TDS, defined based on the batch size, should be randomly tested after they have been placed in their primary packaging material.
- The sampled TDS should be removed from their packaging and thoroughly visually inspected for leakage.
- Each sampled TDS should then be uniformly wiped with a solvent-moistened swab. Both the backing side and the release liner side of the TDS should be wiped. The inside surface of the pouch should also be wiped. The swab(s) is (are) then extracted and assayed for the drug.
- The product fails if the total amount of drug from the TDS, and the corresponding pouch, exceed the acceptable limit established by the manufacturer.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Margareth R.C. Marques, Ph.D.
Senior Scientific Liaison 1-301-816-8106 |
(GCDF2010) General Chapters - Dosage Forms |
USP35–NF30 Page 37
Pharmacopeial Forum: Volume No. 36(6) Page 1647