INTRODUCTION
This general chapter applies to existing drug substances, excipients, and products. All substances and products are subject to relevant control of solvents likely to be present in a substance or product.
Where the limits to be applied comply with those given below, tests for residual solvents are not generally mentioned in specific monographs, because the solvents employed may vary from one manufacturer to another.
The objective of this general chapter is to provide acceptable amounts of residual solvents in pharmaceuticals for the safety of the patient. The chapter recommends the use of less toxic solvents and describes levels considered to be toxicologically acceptable for some residual solvents.
For pharmacopeial purposes, residual solvents in pharmaceuticals are defined as organic volatile chemicals that are used or produced in the manufacture of drug substances or excipients, or in the preparation of drug products. The residual solvents are not completely removed by practical manufacturing techniques. Appropriate selection of the solvent for the synthesis of a drug substance or an excipient may enhance the yield, or determine characteristics such as crystal form, purity, and solubility. Therefore, the solvent may sometimes be a critical element in the synthetic process. This general chapter does not address solvents deliberately used as excipients, nor does it address solvates. However, the content of solvents in such products should be evaluated and justified.
Because residual solvents do not provide therapeutic benefit, they should be removed, to the extent possible, to meet ingredient and product specifications, good manufacturing practices, or other quality-based requirements. Drug products should contain no higher levels of residual solvents than can be supported by safety data. Solvents that are known to cause unacceptable toxicities (Class 1, Table 1) should be avoided in the production of drug substances, excipients, or drug products unless their use can be strongly justified in a risk-benefit assessment. Solvents associated with less severe toxicity (Class 2, Table 2) should be limited in order to protect patients from potential adverse effects. Ideally, less toxic solvents (Class 3, Table 3) should be used where practical. The complete list of solvents included in this general chapter is given in Appendix 1. These tables and the list are not exhaustive. For the purposes of this Pharmacopeia, when a manufacturer has received approval from a competent regulatory authority for the use of a new solvent not currently listed in this general chapter, it is the responsibility of that manufacturer to notify the USP regarding the identity of this solvent, the approved residual solvent limit in the article, and the appropriate test procedure for this residual solvent in the article. The USP will then address this topic in the individual monograph. When a new solvent has been approved through the ICH process, this new solvent will be added to the appropriate list in this general chapter. At that time, consideration will be given for removal of the specific solvent test requirement in the individual monograph.
Testing of drug substances, excipients, and drug products for residual solvents should be performed when production or purification processes are known to result in the presence of such residual solvents. It is only necessary to test for residual solvents that are used or produced in the manufacture or purification of drug substances, excipients, or products.
Although manufacturers may choose to test the drug product, a cumulative procedure may be used to calculate the residual solvent levels in the drug product from the levels in the ingredients used to produce the drug product. If the calculation results in a level equal to or below that provided in this general chapter, no testing of the drug product for residual solvents need be considered. If, however, the calculated level is above the recommended level, the drug product should be tested to ascertain whether the formulation process has reduced the relevant solvent level to within the acceptable amount. A drug product should also be tested if a residual solvent is used during its manufacture.
For the purposes of this Pharmacopeia, when a manufacturer has received approval from a competent regulatory authority for a higher level of residual solvent, it is the responsibility of that manufacturer to notify the USP regarding the identity of this solvent and the approved residual solvent limit in the article. The USP will then address this topic in the individual monograph.
See Appendix 2 for additional background information related to residual solvents.
CLASSIFICATION OF RESIDUAL SOLVENTS BY RISK ASSESSMENT
The term tolerable daily intake (TDI) is used by the International Program on Chemical Safety (IPCS) to describe exposure limits of toxic chemicals, and the term acceptable daily intake (ADI) is used by the World Health Organization (WHO) and other national and international health authorities and institutes. The term permitted daily exposure (PDE) is defined as a pharmaceutically acceptable intake of residual solvents to avoid confusion of differing values for ADIs of the same substance.
Residual solvents assessed in this general chapter are listed in Appendix 1 by common names and structures. They were evaluated for their possible risk to human health and placed into one of three classes as follows:
| Residual Solvent Class | Assessment |
|---|---|
| Class 1 | Solvents to be avoided |
| Known human carcinogens | |
| Strongly suspected human carcinogens | |
| Environmental hazards | |
| Class 2 | Solvents to be limited |
| Nongenotoxic animal carcinogens or possible causative agents of other irreversible toxicity, such as neurotoxicity or teratogenicity | |
| Solvents suspected of other significant but reversible toxicities | |
| Class 3 | Solvents with low toxic potential |
| Solvents with low toxic potential to humans; no health-based exposure limit is needed [Note—Class 3 residual solvents have PDEs of 50 mg or more per day.* ] | |
|
*
For residual solvents with PDEs of more than 50 mg per day, see the discussion in the section Class 3 under Limits of Residual Solvents.
|
|
METHODS FOR ESTABLISHING EXPOSURE LIMITS
The method used to establish PDEs for residual solvents is presented in Appendix 3.
For articles that are designated “for veterinary use only”, higher levels for the PDE and concentration limit may be justified in exceptional cases based upon the actual daily dose, actual target species, and relevant toxicological data and considering consumer safety impact. For the purpose of this Pharmacopeia, when a manufacturer has received approval from a competent regulatory authority for a higher limit, it is the responsibility of that manufacturer to notify the USP regarding the approved residual solvent limit in the article and the justification. The USP will then address this topic in the individual monograph.
OPTIONS FOR DESCRIBING LIMITS OF CLASS 2 RESIDUAL SOLVENTS
Two options are available when setting limits for Class 2 residual solvents.
Option 1
The concentration limits in ppm stated in Table 2 are used. They were calculated using the equation below by assuming a product weight of 10 g administered daily.
Concentration (ppm) = (1000 µg/mg x PDE)/dose
Here, PDE is given in terms of mg per day, and dose is given in g per day.
These limits are considered acceptable for all drug substances, excipients, and drug products. Therefore, this option may be applied if the daily dose is not known or fixed. If all drug substances and excipients in a formulation meet the limits given in Option 1, these components may be used in any proportion. No further calculation is necessary, provided that the daily dose does not exceed 10 g. Products that are administered in doses greater than 10 g per day are to be considered under Option 2.
Option 2
It is not necessary for each component of the drug product to comply with the limits given in Option 1. The PDE in terms of mg per day as stated in Table 2 can be used with the known maximum daily dose and the equation above to determine the concentration of residual solvent allowed in a drug product. Such limits are considered acceptable, provided that it has been demonstrated that the residual solvent has been reduced to the practical minimum. The limits should be realistic in relation to analytical precision, manufacturing capability, and reasonable variation in the manufacturing process. The limits should also reflect contemporary manufacturing standards.
Option 2 may be applied by adding the amounts of a residual solvent present in each of the components of the drug product. The sum of the amounts of solvent per day should be less than that given by the PDE.
Consider an example of the application of Option 1 and Option 2 to acetonitrile concentration in a drug product. The permitted daily exposure to acetonitrile is 4.1 mg per day; thus, the Option 1 limit is 410 ppm. The maximum administered daily weight of a drug product is 5.0 g, and the drug product contains two excipients. The composition of the drug product and the calculated maximum content of residual acetonitrile are given in the following table.
| Component | Amount in Formulation (g) |
Acetonitrile Content (ppm) |
Daily Exposure (mg) |
|---|---|---|---|
| Drug substance | 0.3 | 800 | 0.24 |
| Excipient 1 | 0.9 | 400 | 0.36 |
| Excipient 2 | 3.8 | 800 | 3.04 |
| Drug product | 5.0 | 728 | 3.64 |
Excipient 1 meets the Option 1 limit, but the drug substance, excipient 2, and drug product do not meet the Option 1 limit. Nevertheless, the drug product meets the Option 2 limit of 4.1 mg per day and thus conforms to the acceptance criteria in this general chapter.
Consider another example, using acetonitrile as the residual solvent. The maximum administered daily weight of a drug product is 5.0 g, and the drug product contains two excipients. The composition of the drug product and the calculated maximum content of residual acetonitrile are given in the following table.
| Component | Amount in Formulation (g) |
Acetonitrile Content (ppm) |
Daily Exposure (mg) |
|---|---|---|---|
| Drug substance | 0.3 | 800 | 0.24 |
| Excipient 1 | 0.9 | 2000 | 1.80 |
| Excipient 2 | 3.8 | 800 | 3.04 |
| Drug product | 5.0 | 1016 | 5.08 |
In this example, the drug product meets neither the Option 1 nor the Option 2 limit according to this summation. The manufacturer could test the drug product to determine whether the formulation process reduced the level of acetonitrile. If the level of acetonitrile was not reduced to the allowed limit during formulation, the product fails to meet the solvent limits as described in this chapter, and the manufacturer of the drug product should take other steps to reduce the amount of acetonitrile in the drug product. In some instances the manufacturer may have received approval from a competent regulatory authority for such a higher level of residual solvent. If this is the case, it is the responsibility of that manufacturer to notify the USP regarding the identity of this solvent and the approved residual solvent limit in the article. The USP will then address this topic in the individual monograph.
ANALYTICAL PROCEDURES
Residual solvents are typically determined using chromatographic techniques such as gas chromatography. Compendial methods for testing for residual solvent content are described under the section Identification, Control, and Quantification of Residual Solvents in this general chapter. The General Notices discuss the use of other methods in special circumstances (see 6.30. Alternative and Harmonized Methods and Procedures). If Class 3 solvents are present, a nonspecific method such as loss on drying may be used.
REPORTING LEVELS OF RESIDUAL SOLVENTS
Manufacturers of pharmaceutical products need certain information about the content of residual solvents in drug substances or excipients in order to meet the criteria of this general chapter. The following statements are given as acceptable examples of the information that could be provided from a supplier of drug substances or excipients to a pharmaceutical manufacturer. The supplier might choose one of the following as appropriate:
- Only Class 3 solvents are likely to be present. Loss on drying is less than 0.5%.
- Only Class 2 solvents X, Y, ... are likely to be present. All are below the Option 1 limit. (Here the supplier would name the Class 2 solvents represented by X, Y, ...)
- Only Class 2 solvents X, Y, ... and Class 3 solvents are likely to be present. Residual Class 2 solvents are below the Option 1 limit and residual Class 3 solvents are below 0.5%.
The phrase “likely to be present” as used in the above examples refers to the solvent used or produced in the final manufacturing step and to solvents that are used or produced in earlier manufacturing steps and not removed consistently by a validated process.
If Class 1 solvents are likely to be present, they should be identified and quantified. If solvents of Class 2 or 3 are present at greater than their Option 1 limits or 0.5%, respectively, they should be identified and quantified.
Change to read:
LIMITS OF RESIDUAL SOLVENTS
Class 1 (solvents to be avoided)
Class 1 residual solvents (Table 1) should not be employed in the manufacture of drug substances, excipients, and drug products because of the unacceptable toxicities or deleterious environmental effects of these residual solvents. However, if their use in order to produce a medicinal product with a significant therapeutic advance is unavoidable, their levels should be restricted as shown in Table 1, unless otherwise stated in the individual monograph. The solvent 1,1,1-trichloroethane is included in Table 1 because it is an environmental hazard. The stated limit of 1500 ppm is based on a review of safety data.
When Class 1 residual solvents are used or produced in the manufacture or purification of a drug substance, excipient, or drug product and are not removed by the process, these solvents should be identified and quantified. The procedures described in the section Identification, Control, and Quantification of Residual Solvents in this general chapter are to be applied wherever possible. Otherwise an appropriate validated procedure is to be employed.
Table 1. Class 1 Residual Solvents
(solvents that should be avoided)
(solvents that should be avoided)
| Solvent | Concentration Limit (ppm) |
Concern |
|---|---|---|
| Benzene | 2 | Carcinogen |
| Carbon tetrachloride | 4 | Toxic and environmental hazard |
| 1,2-Dichloroethane | 5 | Toxic |
| 1,1-Dichloroethene | 8 | Toxic |
| 1,1,1-Trichloroethane | 1500 | Environmental hazard |
Class 2
Class 2 residual solvents (Table 2) should be limited in drug substances, excipients, and drug products because of the inherent toxicities of the residual solvents. PDEs are given to the nearest 0.1 mg per day, and concentrations are given to the nearest 10 ppm. The stated values do not reflect the necessary analytical precision of the determination procedure. Precision should be determined as part of the procedure validation.
If Class 2 residual solvents are present at greater than their Option 1 limits, they should be identified and quantified. The procedures described in the section Identification, Control, and Quantification of Residual Solvents in this general chapter are to be applied wherever possible. Otherwise an appropriate validated procedure is to be employed.
[Note—The following Class 2 residual solvents are not readily detected by the headspace injection conditions described in the section Identification, Control, and Quantification of Residual Solvents in this general chapter: formamide, 2-ethoxyethanol, 2-methoxyethanol, ethylene glycol, N-methylpyrrolidone, and sulfolane. Other appropriate validated procedures are to be employed for the quantification of these residual solvents. Such procedures shall be submitted to the USP for review and possible inclusion in the relevant individual monograph. In addition, USP Residual Solvent Class 2—Mixture C RS can be used to develop an alternative procedure. ]
Table 2. Class 2 Residual Solvents
| Solvent | PDE (mg/day) |
Concentration Limit (ppm) |
|---|---|---|
| Acetonitrile | 4.1 | 410 |
| Chlorobenzene | 3.6 | 360 |
| Chloroform | 0.6 | 60 |
| Cyclohexane | 38.8 | 3880 |
| 1,2-Dichloroethene | 18.7 | 1870 |
| 1,2-Dimethoxyethane | 1.0 | 100 |
| N,N-Dimethylacetamide | 10.9 | 1090 |
| N,N-Dimethylformamide | 8.8 | 880 |
| 1,4-Dioxane | 3.8 | 380 |
| 2-Ethoxyethanol | 1.6 | 160 |
| Ethylene glycol | 6.2 | 620 |
| Formamide | 2.2 | 220 |
| Hexane | 2.9 | 290 |
| Methanol | 30.0 | 3000 |
| 2-Methoxyethanol | 0.5 | 50 |
| Methylbutylketone | 0.5 | 50 |
| Methylcyclohexane | 11.8 | 1180 |
| Methylene chloride | 6.0 | 600 |
| N-Methylpyrrolidone | 5.3 | 530 |
| Nitromethane | 0.5 | 50 |
| Pyridine | 2.0 | 200 |
| Sulfolane | 1.6 | 160 |
| Tetrahydrofuran | 7.2 | 720 |
| Tetralin | 1.0 | 100 |
| Toluene | 8.9 | 890 |
| Trichloroethylene | 0.8 | 80 |
| Xylene* | 21.7 | 2170 |
|
*
Usually 60% m-xylene, 14% p-xylene, 9% o-xylene with 17% ethyl benzene.
|
||
Class 3
Class 3 residual solvents (Table 3) may be regarded as less toxic and of lower risk to human health than Class 1 and Class 2 residual solvents. Class 3 includes no solvent known as a human health hazard at levels normally accepted in pharmaceuticals. However, there are no long-term toxicity or carcinogenicity studies for many of the residual solvents in Class 3. Available data indicate that they are less toxic in acute or short-term studies and negative in genotoxicity studies.
It is considered that amounts of these residual solvents of 50 mg per day or less (corresponding to 5000 ppm or 0.5% under Option 1) would be acceptable without justification. Higher amounts may also be acceptable, provided that they are realistic in relation to manufacturing capability and good manufacturing practice. For the purposes of this Pharmacopeia, when a manufacturer has received approval from a competent regulatory authority for such a higher level of residual solvent, it is the responsibility of that manufacturer to notify the USP regarding the identity of this solvent and the approved residual solvent limit in the article. The USP will then address this topic in the individual monograph. If a Class 3 solvent limit in an individual monograph is greater than 50 mg per day, that residual solvent should be identified and quantified. The procedures described in the section Identification, Control, and Quantification of Residual Solvents in this general chapter, with appropriate modifications to the standard solutions, are to be applied wherever possible. Otherwise an appropriate validated procedure is to be employed.
Table 3. Class 3 Residual Solvents
(limited by GMP or other quality-based requirements in drug substances, excipients, and drug products)
(limited by GMP or other quality-based requirements in drug substances, excipients, and drug products)
| Acetic acid | Heptane |
| Acetone | Isobutyl acetate |
| Anisole | Isopropyl acetate |
| 1-Butanol | Methyl acetate |
| 2-Butanol | 3-Methyl-1-butanol |
| Butyl acetate | Methylethylketone |
| tert-Butylmethyl ether | Methylisobutylketone |
| Cumene | 2-Methyl-1-propanol |
| Dimethyl sulfoxide | Pentane |
| Ethanol | 1-Pentanol |
| Ethyl acetate | 1-Propanol |
| Ethyl ether | 2-Propanol |
| Ethyl formate | Propyl acetate |
| Formic acid |
Other Residual Solvents
The residual solvents listed in Table 4 may also be of interest to manufacturers of drug substances, excipients, or drug products. However, no adequate toxicological data on which to base a PDE was found.
Table 4. Other Residual Solvents
(for which no adequate toxicological data was found)
(for which no adequate toxicological data was found)
| 1,1-Diethoxypropane | Methyl isopropyl ketone |
| 1,1-Dimethoxymethane | Methyltetrahydrofuran |
| 2,2-Dimethoxypropane | Solvent hexane |
| Isooctane | Trichloroacetic acid |
| Isopropyl ether | Trifluoroacetic acid |
IDENTIFICATION, CONTROL, AND QUANTIFICATION OF RESIDUAL SOLVENTS
Whenever possible, the substance under test needs to be dissolved to release the residual solvent. Because the USP deals with drug products, as well as active ingredients and excipients, it may be acceptable that in some cases, some of the components of the formulation will not dissolve completely. In those cases, the drug product may first need to be pulverized into a fine powder so that any residual solvent that may be present can be released. This operation should be performed as fast as possible to prevent the loss of volatile solvents during the procedure.
Note—The organic-free water specified in the following procedures produces no significantly interfering peaks when chromatographed.
Class 1 and Class 2 Residual Solvents
The following procedures are useful to identify and quantify residual solvents when the information regarding which solvents are likely to be present in the material is not available. When the information about the presence of specific residual solvents is available, only Procedure C is needed to quantify the amount of residual solvents present. A flow diagram for the application of the residual solvent limit tests is shown in Figure 1.
+'&img=../../images/v35300/c467-f1-usp33.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 1. Diagram relating to the identification of residual solvents and the application of limit tests.
water-soluble articles
Procedure A—
Class 1 Standard Stock Solution—
[note—When transferring solutions, place the tip of the pipet just below the surface of the liquid, and mix. ] Transfer 1.0 mL of USP Class 1 Residual Solvents Mixture RS to a 100-mL volumetric flask, previously filled with about 9 mL of dimethyl sulfoxide, dilute with water to volume, and mix. Transfer 1.0 mL of this solution to a 100-mL volumetric flask, previously filled with about 50 mL of water, dilute with water to volume, and mix. Transfer 10 mL of this solution to a 100-mL volumetric flask, previously filled with about 50 mL of water, dilute with water to volume, and mix.
Class 1 Standard Solution—
Transfer 1.0 mL of Class 1 Standard Stock Solution to an appropriate headspace vial containing 5.0 mL of water (place the tip of the pipet just below the surface of the liquid for dispensing), apply the stopper, cap, and mix.
Class 2 Standard Stock Solutions—
Transfer 1.0 mL of USP Residual Solvents Class 2—Mixture A RS to a 100-mL volumetric flask, dilute with water to volume, and mix. This is Class 2 Standard Stock Solution A. Transfer 1.0 mL of USP Residual Solvents Class 2—Mixture B RS to a 100-mL volumetric flask, dilute with water to volume, and mix. This is Class 2 Standard Stock Solution B.
Class 2 Mixture A Standard Solution—
Transfer 1.0 mL of Class 2 Standard Stock Solution A to an appropriate headspace vial, add 5.0 mL of water, apply the stopper, cap, and mix.
Class 2 Mixture B Standard Solution—
Transfer 5.0 mL of Class 2 Standard Stock Solution B to an appropriate headspace vial, add 1.0 mL of water, apply the stopper, cap, and mix.
Test Stock Solution—
Transfer about 250 mg of the article under test, accurately weighed, to a 25-mL volumetric flask, dissolve in and dilute with water to volume, and mix.
Test Solution—
Transfer 5.0 mL of Test Stock Solution to an appropriate headspace vial, add 1.0 mL of water, apply the stopper, cap, and mix.
Class 1 System Suitability Solution—
Transfer 1.0 mL of Class 1 Standard Stock Solution to an appropriate headspace vial, add 5.0 mL of Test Stock Solution, apply the stopper, cap, and mix.
Chromatographic System (see Chromatography  621
621 )—
The gas chromatograph is equipped with a flame-ionization detector and a 0.32-mm × 30-m fused-silica column coated with a 1.8-µm layer of phase G43 or a 0.53-mm × 30-m wide-bore column coated with a 3.0-µm layer of phase G43. The carrier gas is nitrogen or helium with a linear velocity of about 35 cm per second, and a split ratio of 1:5. [Note—The split ratio can be modified in order to optimize sensitivity. ] The column temperature is maintained at 40
)—
The gas chromatograph is equipped with a flame-ionization detector and a 0.32-mm × 30-m fused-silica column coated with a 1.8-µm layer of phase G43 or a 0.53-mm × 30-m wide-bore column coated with a 3.0-µm layer of phase G43. The carrier gas is nitrogen or helium with a linear velocity of about 35 cm per second, and a split ratio of 1:5. [Note—The split ratio can be modified in order to optimize sensitivity. ] The column temperature is maintained at 40 for 20 minutes, then raised at a rate of 10 per minute to 240, and maintained at 240 for 20 minutes. The injection port and detector temperatures are maintained at 140 and 250, respectively. Chromatograph the Class 1 Standard Solution, Class 1 System Suitability Solution, and Class 2 Mixture A Standard Solution, and record the peak responses as directed for Procedure: the signal-to-noise ratio of 1,1,1-trichloroethane in the Class 1 Standard Solution is not less than 5; the signal-to-noise ratio of each peak in the Class 1 System Suitability Solution is not less than 3; and the resolution, R, between acetonitrile and methylene chloride in the Class 2 Mixture A Standard Solution is not less than 1.0.
for 20 minutes, then raised at a rate of 10 per minute to 240, and maintained at 240 for 20 minutes. The injection port and detector temperatures are maintained at 140 and 250, respectively. Chromatograph the Class 1 Standard Solution, Class 1 System Suitability Solution, and Class 2 Mixture A Standard Solution, and record the peak responses as directed for Procedure: the signal-to-noise ratio of 1,1,1-trichloroethane in the Class 1 Standard Solution is not less than 5; the signal-to-noise ratio of each peak in the Class 1 System Suitability Solution is not less than 3; and the resolution, R, between acetonitrile and methylene chloride in the Class 2 Mixture A Standard Solution is not less than 1.0.
Procedure—
[Note—It is recommended to increase the temperature of the transfer line between runs to eliminate any potential condensation of solvents. ] Separately inject (following one of the headspace operating parameter sets described in Table 5) equal volumes of headspace (about 1.0 mL) of the Class 1 Standard Solution, Class 2 Mixture A Standard Solution, Class 2 Mixture B Standard Solution, and Test Solution into the chromatograph, record the chromatograms, and measure the responses for the major peaks. If a peak response of any peak, other than a peak for 1,1,1-trichloroethane, in the Test Solution is greater than or equal to a corresponding peak in either the Class 1 Standard Solution or either of the two Class 2 Mixture Standard Solutions, or a peak response of 1,1,1-trichloroethane is greater than or equal to 150 times the peak response corresponding to 1,1,1-trichloroethane in the Class 1 Standard Solution, proceed to Procedure B to verify the identity of the peak; otherwise the article meets the requirements of this test.
Table 5. Headspace Operating Parameters
| Headspace Operating Parameter Sets |
|||
|---|---|---|---|
| 1 | 2 | 3 | |
| Equilibration temperature ( |
80 | 105 | 80 |
| Equilibration time (min) | 60 | 45 | 45 |
| Transfer-line temperature ( |
85 | 110 | 105 |
| Syringe temperature ( |
80–90 | 105–115 | 80–90 |
| Carrier gas: nitrogen or helium at an appropriate pressure | |||
| Pressurization time(s) (if appropriate) | |||
| Injection volume (mL)* | 1 | 1 | 1 |
|
*
Or follow the instrument manufacturer's recommendations, as long as the method criteria are met. Injecting less than this amount is allowed as long as adequate sensitivity is achieved.
|
|||
Procedure B—
Class 1 Standard Stock Solution, Class 1 Standard Solution, Class 2 Standard Stock Solutions, Class 2 Mixture A Standard Solution, Class 2 Mixture B Standard Solution, Test Stock Solution, Test Solution, and Class 1 System Suitability Solution—
Prepare as directed for Procedure A.
Chromatographic System (see Chromatography 621)—
The gas chromatograph is equipped with a flame-ionization detector and a 0.32-mm × 30-m fused-silica column coated with a 0.25-µm layer of phase G16 or a 0.53-mm × 30-m wide-bore column coated with a 0.25-µm layer of phase G16. The carrier gas is nitrogen or helium with a linear velocity of about 35 cm per second and a split ratio of 1:5. [Note—The split ratio can be modified in order to optimize sensitivity. ] The column temperature is maintained at 50 for 20 minutes, then raised at a rate of 6 per minute to 165, and maintained at 165 for 20 minutes. The injection port and detector temperatures are maintained at 140 and 250, respectively. Chromatograph the Class 1 Standard Solution and the Class 1 System Suitability Solution, and record the peak responses as directed for Procedure: the signal-to-noise ratio of benzene in the Class 1 Standard Solution is not less than 5; the signal-to-noise ratio of each peak in the Class 1 System Suitability Solution is not less than 3; and the resolution, R, between acetonitrile and cis-dichloroethene in the Class 2 Mixture A Standard Solution is not less than 1.0.
Procedure—
[Note—It is recommended to increase the temperature of the transfer line between runs to eliminate any potential condensation of solvents. ] Separately inject (following one of the headspace operating parameter sets described in Table 5) equal volumes of headspace (about 1.0 mL) of the Class 1 Standard Solution, the Class 2 Mixture A Standard Solution, the Class 2 Mixture B Standard Solution, and the Test Solution into the chromatograph, record the chromatograms, and measure the responses for the major peaks. If the peak response(s) in the Test Solution of the peak(s) identified in Procedure A is/are greater than or equal to a corresponding peak(s) in either the Class 1 Standard Solution or either of the two Class 2 Mixture Standard Solutions, proceed to Procedure C to quantify the peak(s); otherwise the article meets the requirements of this test.
Procedure C—
Class 1 Standard Stock Solution, Class 1 Standard Solution, Class 2 Standard Stock Solution A, Class 2 Mixture A Standard Solution, Test Stock Solution, Test Solution, and Class 1 System Suitability Solution—
Prepare as directed for Procedure A.
Standard Stock Solution—
[note—Prepare a separate Standard Stock Solution for each peak identified and verified by Procedures A and B. For the Class 1 solvents other than 1,1,1-trichloroethane, prepare the first dilution as directed for the first dilution under Class 1 Standard Stock Solution in Procedure A. ] Transfer an accurately measured volume of each individual USP Reference Standard corresponding to each residual solvent peak identified and verified by Procedures A and B to a suitable container, and dilute quantitatively, and stepwise if necessary, with water to obtain a solution having a final concentration of 1/20 of the value stated in Table 1 or 2 (under Concentration Limit).
Standard Solution—
Transfer 1.0 mL of this solution to an appropriate headspace vial, add 5.0 mL of water, apply the stopper, cap, and mix.
Spiked Test Solution—
[Note—Prepare a separate Spiked Test Solution for each peak identified and verified by Procedures A and B. ] Transfer 5.0 mL of Test Stock Solution to an appropriate headspace vial, add 1.0 mL of the Standard Stock Solution, apply the stopper, cap, and mix.
Chromatographic System (see Chromatography 621)—
[Note—If the results of the chromatography from Procedure A are found to be inferior to those found with Procedure B, the Chromatographic System from Procedure B may be substituted. ] The gas chromatograph is equipped with a flame-ionization detector and a 0.32-mm × 30-m fused-silica column coated with a 1.8-µm layer of phase G43 or a 0.53-mm × 30-m wide-bore column coated with a 3.0-µm layer of phase G43. The carrier gas is nitrogen or helium with a linear velocity of about 35 cm per second, and a split ratio of 1:5. [Note—The split ratio can be modified in order to optimize sensitivity. ] The column temperature is maintained at 40 for 20 minutes, then raised at a rate of 10 per minute to 240, and maintained at 240 for 20 minutes. The injection port and detector temperatures are maintained at 140 and 250, respectively. Chromatograph the Class 1 Standard Solution, the Class 1 System Suitability Solution, and the Class 2 Mixture A Standard Solution, and record the peak responses as directed for Procedure: the signal-to-noise ratio of 1,1,1-trichloroethane in the Class 1 Standard Solution is not less than 5; the signal-to-noise ratio of each peak in the Class 1 System Suitability Solution is not less than 3; and the resolution, R, between acetonitrile and methylene chloride in the Class 2 Mixture A Standard Solution is not less than 1.0.
Procedure—
[Note—It is recommended to increase the temperature of the transfer line between runs to eliminate any potential condensation of solvents. ] Separately inject (following one of the headspace operating parameters described in Table 5) equal volumes of headspace (about 1.0 mL) of the Standard Solution, the Test Solution, and the Spiked Test Solution into the chromatograph, record the chromatograms, and measure the responses for the major peaks. Calculate the amount, in ppm, of each residual solvent found in the article under test by the formula:
5(C/W)[rU /(rST 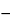 rU)]
rU)]
in which C is the concentration, in µg per mL, of the appropriate USP Reference Standard in the Standard Stock Solution; W is the weight, in g, of the article under test taken to prepare the Test Stock Solution; and rU and rST are the peak responses of each residual solvent obtained from the Test Solution and the Spiked Test Solution, respectively.
water-insoluble articles
Procedure A—
[Note—Dimethyl sulfoxide may be substituted as an alternative solvent to dimethylformamide. ]
Class 1 Standard Stock Solution—
Transfer 1.0 mL of USP Class 1 Residual Solvents Mixture RS to a 100-mL volumetric flask previously filled with about 80 mL of dimethylformamide, dilute with dimethylformamide to volume, and mix. Transfer 1.0 mL of this solution to a 100-mL volumetric flask, previously filled with about 80 mL of dimethylformamide, dilute with dimethylformamide to volume, and mix (reserve a portion of this solution for the Class 1 System Suitability Solution). Transfer 1.0 mL of this solution to a 10-mL volumetric flask, dilute with dimethylformamide to volume, and mix.
Class 1 Standard Solution—
Transfer 1.0 mL of Class 1 Standard Stock Solution to an appropriate headspace vial, containing 5.0 mL of water, apply the stopper, cap, and mix.
Class 2 Standard Stock Solutions—
Transfer 1.0 mL of USP Residual Solvents Class 2—Mixture A RS to a 100-mL volumetric flask, previously filled with about 80 mL of dimethylformamide, dilute with dimethylformamide to volume, and mix. This is Class 2 Standard Stock Solution A. Transfer 0.5 mL of USP Residual Solvents Class 2—Mixture B RS to a 10-mL volumetric flask, dilute with dimethylformamide to volume, and mix. This is Class 2 Standard Stock Solution B.
Class 2 Mixture A Standard Solution—
Transfer 1.0 mL of Class 2 Standard Stock Solution A to an appropriate headspace vial containing 5.0 mL of water, apply the stopper, cap, and mix.
Class 2 Mixture B Standard Solution—
Transfer 1.0 mL of Class 2 Standard Stock Solution B to an appropriate headspace vial containing 5.0 mL of water, apply the stopper, cap, and mix.
Test Stock Solution—
Transfer about 500 mg of the article under test, accurately weighed, to a 10-mL volumetric flask, dissolve in and dilute with dimethylformamide to volume, and mix.
Test Solution—
Transfer 1.0 mL of Test Stock Solution to an appropriate headspace vial, containing 5.0 mL of water, apply the stopper, cap, and mix.
Class 1 System Suitability Solution—
Mix 5 mL of Test Stock Solution with 0.5 mL of the intermediate dilution reserved from Class 1 Standard Stock Solution. Transfer 1.0 mL of this solution to an appropriate headspace vial, containing 5.0 mL of water, apply the stopper, cap, and mix.
Chromatographic System (see Chromatography 621—
The gas chromatograph is equipped with a flame-ionization detector and a 0.53-mm × 30-m wide-bore column coated with a 3.0-µm layer of phase G43. The carrier gas is helium with a linear velocity of about 35 cm per second and a split ratio of 1:3. [Note—The split ratio can be modified in order to optimize sensitivity. ] The column temperature is maintained at 40 for 20 minutes, then raised at a rate of 10 per minute to 240, and maintained at 240 for 20 minutes. The injection port and detector temperatures are maintained at 140 and 250, respectively. Chromatograph the Class 1 Standard Solution, Class 1 System Suitability Solution, and Class 2 Mixture A Standard Solution, and record the peak responses as directed for Procedure: the signal-to-noise ratio of 1,1,1-trichloroethane in the Class 1 Standard Solution is not less than 5; the signal-to-noise ratio of each peak in the Class 1 System Suitability Solution is not less than 3; and the resolution, R, between acetonitrile and methylene chloride in the Class 2 Mixture A Standard Solution is not less than 1.0.
Procedure—
[Note—It is recommended to increase the temperature of the transfer line between runs to eliminate any potential condensation of solvents. ] Separately inject (use headspace operating parameters in column 3 of Table 5 with a vial pressure of 10 psi) equal volumes of headspace (about 1.0 mL) of the Class 1 Standard Solution, Class 2 Mixture A Standard Solution, Class 2 Mixture B Standard Solution, and Test Solution into the chromatograph, record the chromatograms, and measure the responses for the major peaks. If a peak response of any peak, other than a peak for 1,1,1-trichloroethane, in the Test Solution is greater than or equal to a corresponding peak in either the Class 1 Standard Solution or either of the two Class 2 Mixture Standard Solutions, or a peak response of 1,1,1-trichloroethane is greater than or equal to 150 times the peak response corresponding to 1,1,1-trichloroethane in the Class 1 Standard Solution, proceed to Procedure B to verify the identity of the peak; otherwise the article meets the requirements of this test.
Procedure B—
Class 1 Standard Stock Solution, Class 1 Standard Solution, Class 1 System Suitability Solution, Class 2 Standard Stock Solutions, Class 2 Mixture A Standard Solution, and Class 2 Mixture B Standard Solution, Test Stock Solution, and Test Solution—
Proceed as directed for Procedure A.
Chromatographic System—
Proceed as directed for Procedure B under Water-Soluble Articles with a split ratio of 1:3.
[Note—The split ratio can be modified in order to optimize sensitivity. ]
Procedure—
[Note—It is recommended to increase the temperature of the transfer line between runs to eliminate any potential condensation of solvents. ] Separately inject (use headspace operating parameters in column 3 of Table 5 with a vial pressure of 10 psi) equal volumes of headspace (about 1.0 mL) of the Class 1 Standard Solution, Class 2 Mixture A Standard Solution, Class 2 Mixture B Standard Solution, and Test Solution into the chromatograph, record the chromatograms, and measure the responses for the major peaks. If the peak response(s) in Test Solution of the peak(s) identified in Procedure A is/are greater than or equal to a corresponding peak(s) in either the Class 1 Standard Solution or any of the two Class 2 Mixture Standard Solutions, proceed to Procedure C to quantify the peak(s); otherwise the article meets the requirements of this test.
Procedure C—
Class 1 Standard Stock Solution, Class 1 Standard Solution, Class 1 System Suitability Solution, Class 2 Standard Stock Solution A, and Class 2 Mixture A Standard Solution—
Proceed as directed for Procedure A.
Standard Stock Solution—
[note—Prepare a separate Standard Stock Solution for each peak identified and verified by Procedures A and B. For the Class 1 solvents other than 1,1,1-trichloroethane, prepare the first dilution as directed for the first dilution under Class 1 Standard Stock Solution in Procedure A. ] Transfer an accurately measured volume of each individual USP Reference Standard corresponding to each residual solvent peak identified and verified by Procedures A and B to a suitable container, and dilute quantitatively, and stepwise if necessary, with water to obtain a solution having a final concentration of 1/20 of the value stated in Table 1 or Table 2 (under Concentration Limit).
Standard Solution—
Transfer 1.0 mL of the Standard Stock Solution to an appropriate headspace vial, containing 5.0 mL of water, apply the stopper, cap, and mix.
Test Stock Solution—
Proceed as directed for Procedure A.
Test Solution—
Transfer 1.0 mL of the Test Stock Solution to an appropriate headspace vial, containing 5.0 mL of water, apply the stopper, cap, and mix.
Spiked Test Solution—
[Note—Prepare a separate Spiked Test Solution for each peak identified and verified by Procedures A and B. ] Transfer 1.0 mL of Test Stock Solution to an appropriate headspace vial, add 1 mL of Standard Stock Solution and 4.0 mL of water, apply the stopper, cap, and mix.
Chromatographic System—
Proceed as directed for Procedure C under Water-Soluble Articles.
Procedure—
[Note—It is recommended to increase the temperature of the transfer line between runs to eliminate any potential condensation of solvents. ] Separately inject (use headspace operating parameters in column 3 of Table 5 with a vial pressure of 10 psi) equal volumes of headspace (about 1.0 mL) of the Standard Solution, Test Solution, and Spiked Test Solution into the chromatograph, record the chromatograms, and measure the responses for the major peaks. Calculate the amount, in ppm, of each residual solvent found in the article under test by the formula:
10(C/W)[rU/(rST rU)]
in which C is the concentration, in µg per mL, of the appropriate USP Reference Standard in the Standard Stock Solution; W is the weight, in g, of the article under test taken to prepare the Test Stock Solution; and rU and rST are the peak responses of each residual solvent obtained from Test Solution and Spiked Test Solution, respectively.
Class 3 Residual Solvents
If Class 3 solvents are present, the level of residual solvents may be determined as directed under Loss on Drying 731 when the monograph for the article under test contains a loss on drying procedure specifying an upper limit of no more than 0.5% (per Option 1 in this general chapter), or a specific determination of the solvent may be made. If there is no loss on drying procedure in the monograph for the article under test or if a Class 3 solvent limit in an individual monograph is greater than 50 mg per day (corresponding to 5000 ppm or 0.5% under Option 1), the individual Class 3 residual solvent or solvents present in the article under test should be identified and quantified, and the procedures as described above, with appropriate modifications to the standard solutions, are to be applied wherever possible. Otherwise an appropriate validated procedure is to be employed. USP Reference Standards, where available, should be used in these procedures.
GLOSSARY
Acceptable daily intake (ADI): The maximum acceptable intake of toxic chemicals per day. This term is used by the World Health Organization (WHO).
Genotoxic carcinogens: Carcinogens that produce cancer by affecting genes or chromosomes.
Lowest-observed-effect level (LOEL): The lowest dose of a substance in a study or group of studies that produces biologically significant increases in frequency or severity of any effects in exposed humans or animals.
Modifying factor: A factor determined by professional judgment of a toxicologist and applied to bioassay data so that the data can be safely related to humans.
Neurotoxicity: The ability of a substance to cause adverse effects on the nervous system.
No-observed-effect level (NOEL): The highest dose of a substance at which there are no biologically significant increases in frequency or severity of any effects in exposed humans or animals.
Permitted daily exposure (PDE): The maximum acceptable intake per day of a residual solvent in pharmaceutical products.
Reversible toxicity: The occurrence of harmful effects that are caused by a substance and that disappear after exposure to the substance ends.
Strongly suspected human carcinogen: A substance for which there is no epidemiological evidence of carcinogenesis but for which there are positive genotoxicity data and clear evidence of carcinogenesis in rodents.
Teratogenicity: The occurrence of structural malformations in a developing fetus when a substance is administered during pregnancy.
Tolerable daily intake (TDI): Tolerable daily exposure to toxic chemicals. Term used by the International Program on Chemical Safety (IPCS).
APPENDIX 1. LIST
APPENDIX 1. LIST OF RESIDUAL SOLVENTS INCLUDED IN THIS GENERAL CHAPTER
| Solvent | Other Names | Structure | Class |
|---|---|---|---|
| Acetic acid | Ethanoic acid | CH3COOH | Class 3 |
| Acetone | 2-Propanone Propan-2-one |
CH3COCH3 | Class 3 |
| Acetonitrile | CH3CN | Class 2 | |
| Anisole | Methoxybenzene |
+'&img=../../images/v35300/cas-100-66-3.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
|
Class 3 |
| Benzene | Benzol |
+'&img=../../images/v35300/cas-71-43-2.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
|
Class 1 |
| 1-Butanol | n-Butyl alcohol Butan-1-ol |
CH3(CH2)3OH | Class 3 |
| 2-Butanol | sec-Butyl alcohol Butan-2-ol |
CH3CH2CH(OH)CH3 | Class 3 |
| Butyl acetate | Acetic acid butyl ester | CH3COO(CH2)3CH3 | Class 3 |
| tert-Butylmethyl ether | 2-Methoxy-2-methylpropane | (CH3)3COCH3 | Class 3 |
| Carbon tetrachloride | Tetrachloromethane | CCl4 | Class 1 |
| Chlorobenzene |
+'&img=../../images/v35300/cas-108-90-7.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
|
Class 2 | |
| Chloroform | Trichloromethane | CHCl3 | Class 2 |
| Cumene | Isopropylbenzene (1-Methylethyl)benzene |
+'&img=../../images/v35300/cas-98-82-8.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
|
Class 3 |
| Cyclohexane | Hexamethylene |
+'&img=../../images/v35300/cas-110-82-7.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
|
Class 2 |
| 1,2-Dichloroethane | sym-Dichloroethane Ethylene dichloride Ethylene chloride |
CH2ClCH2Cl | Class 1 |
| 1,1-Dichloroethene | 1,1-Dichloroethylene Vinylidene chloride |
H2C=CCl2 | Class 1 |
| 1,2-Dichloroethene | 1,2-Dichloroethylene Acetylene dichloride |
ClHC=CHCl | Class 2 |
| 1,2-Dimethoxyethane | Ethyleneglycol dimethyl ether Monoglyme Dimethyl cellosolve |
H3COCH2CH2OCH3 | Class 2 |
| N,N-Dimethylacetamide | DMA | CH3CON(CH3)2 | Class 2 |
| N,N-Dimethylformamide | DMF | HCON(CH3)2 | Class 2 |
| Dimethyl sulfoxide | Methylsulfinylmethane Methyl sulfoxide DMSO |
(CH3)2SO | Class 3 |
| 1,4-Dioxane | p-Dioxane [1,4]Dioxane |
+'&img=../../images/v35300/cas-123-91-1.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
|
Class 2 |
| Ethanol | Ethyl alcohol | CH3CH2OH | Class 3 |
| 2-Ethoxyethanol | Cellosolve | CH3CH2OCH2CH2OH | Class 2 |
| Ethyl acetate | Acetic acid ethyl ester | CH3COOCH2CH3 | Class 3 |
| Ethylene glycol | 1,2-Dihydroxyethane 1,2-Ethanediol |
HOCH2CH2OH | Class 2 |
| Ethyl ether | Diethyl ether Ethoxyethane 1,1¢-Oxybisethane |
CH3CH2OCH2CH3 | Class 3 |
| Ethyl formate | Formic acid ethyl ester | HCOOCH2CH3 | Class 3 |
| Formamide | Methanamide | HCONH2 | Class 2 |
| Formic acid | HCOOH | Class 3 | |
| Heptane | n-Heptane | CH3(CH2)5CH3 | Class 3 |
| Hexane | n-Hexane | CH3(CH2)4CH3 | Class 2 |
| Isobutyl acetate | Acetic acid isobutyl ester | CH3COOCH2CH(CH3)2 | Class 3 |
| Isopropyl acetate | Acetic acid isopropyl ester | CH3COOCH(CH3)2 | Class 3 |
| Methanol | Methyl alcohol | CH3OH | Class 2 |
| 2-Methoxyethanol | Methyl cellosolve | CH3OCH2CH2OH | Class 2 |
| Methyl acetate | Acetic acid methyl ester | CH3COOCH3 | Class 3 |
| 3-Methyl-1-butanol | Isoamyl alcohol Isopentyl alcohol 3-Methylbutan-1-ol |
(CH3)2CHCH2CH2OH | Class 3 |
| Methylbutylketone | 2-Hexanone Hexan-2-one |
CH3(CH2)3COCH3 | Class 2 |
| Methylcyclohexane | Cyclohexylmethane |
+'&img=../../images/v35300/cas-108-87-2.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
|
Class 2 |
| Methylene chloride | Dichloromethane | CH2Cl2 | Class 2 |
| Methylethylketone | 2-Butanone MEK Butan-2-one |
CH3CH2COCH3 | Class 3 |
| Methyl isobutyl ketone | 4-Methylpentan-2-one 4-Methyl-2-pentanone MIBK |
CH3COCH2CH(CH3)2 | Class 3 |
| 2-Methyl-1-propanol | Isobutyl alcohol 2-Methylpropan-1-ol |
(CH3)2CHCH2OH | Class 3 |
| N-Methylpyrrolidone | 1-Methylpyrrolidin-2-one 1-Methyl-2-pyrrolidinone |
+'&img=../../images/v35300/cas-872-50-4.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
|
Class 2 |
| Nitromethane | CH3NO2 | Class 2 | |
| Pentane | n-Pentane | CH3(CH2)3CH3 | Class 3 |
| 1-Pentanol | Amyl alcohol Pentan-1-ol Pentyl alcohol |
CH3(CH2)3CH2OH | Class 3 |
| 1-Propanol | Propan-1-ol Propyl alcohol |
CH3CH2CH2OH | Class 3 |
| 2-Propanol | Propan-2-ol Isopropyl alcohol |
(CH3)2CHOH | Class 3 |
| Propyl acetate | Acetic acid propyl ester | CH3COOCH2CH2CH3 | Class 3 |
| Pyridine |
+'&img=../../images/v35300/cas-110-86-1.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
|
Class 2 | |
| Sulfolane | Tetrahydrothiophene 1,1-dioxide |
+'&img=../../images/v35300/cas-126-33-0.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
|
Class 2 |
| Tetrahydrofuran | Tetramethylene oxide Oxacyclopentane |
+'&img=../../images/v35300/cas-109-99-9.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
|
Class 2 |
| Tetralin | 1,2,3,4-Tetrahydronaphthalene |
+'&img=../../images/v35300/cas-119-64-2.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
|
Class 2 |
| Toluene | Methylbenzene |
+'&img=../../images/v35300/cas-108-88-3.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
|
Class 2 |
| 1,1,1-Trichloroethane | Methylchloroform | CH3CCl3 | Class 1 |
| Trichloroethylene | 1,1,2-Trichloroethene | HClC=CCl2 | Class 2 |
| Xylene* | Dimethylbenzene Xylol |
+'&img=../../images/v35300/cas-1330-20-7.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
|
Class 2 |
|
*
Usually 60% m-xylene, 14% p-xylene, 9% o-xylene with 17% ethyl benzene.
|
|||
APPENDIX 2. ADDITIONAL BACKGROUND
A2.1. Environmental Regulation of Organic Volatile Solvents
Several of the residual solvents frequently used in the production of pharmaceuticals are listed as toxic chemicals in Environmental Health Criteria (EHC) monographs and in the Integrated Risk Information System (IRIS). The objectives of such groups as the International Programme on Chemical Safety (IPCS), the United States Environmental Protection Agency (EPA), and the United States Food and Drug Administration (FDA) include the determination of acceptable exposure levels. The goal is maintenance of environmental integrity and protection of human health against the possible deleterious effects of chemicals resulting from long-term environmental exposure. The procedures involved in the estimation of maximum safe exposure limits are usually based on long-term studies. When long-term study data are unavailable, shorter-term study data can be used with modification of the approach, such as use of larger safety factors. The approach described therein relates primarily to long-term or lifetime exposure of the general population in the ambient environment (i.e., ambient air, food, drinking water, and other media).
A2.2. Residual Solvents in Pharmaceuticals
Exposure limits in this general chapter are established by referring to methodologies and toxicity data described in EHC and IRIS monographs. However, the following specific assumptions about residual solvents to be used in the synthesis and formulation of pharmaceutical products should be taken into account in establishing exposure limits.
- Patients (not the general population) use pharmaceuticals to treat their diseases or for prophylaxis to prevent infection or disease.
- The assumption of lifetime patient exposure is not necessary for most pharmaceutical products but may be appropriate as a working hypothesis to reduce risk to human health.
- Residual solvents are unavoidable components in pharmaceutical production and will often be a part of medicinal products.
- Residual solvents should not exceed recommended levels except in exceptional circumstances.
- Data from toxicological studies that are used to determine acceptable levels for residual solvents should have been generated using appropriate protocols such as those described, for example, by the Organization for Economic Cooperation and Development (OECD), EPA, and the FDA Red Book.
APPENDIX 3. PROCEDURES FOR ESTABLISHING EXPOSURE LIMITS
The Gaylor-Kodell method of risk assessment (Gaylor, D.W., and Kodell, R.L. Linear Interpolation Algorithm for Low Dose Assessment of Toxic Substance. Journal of Environmental Pathology and Toxicology, 4:305, 1980) is appropriate for Class 1 carcinogenic solvents. Only in cases where reliable carcinogenicity data are available should extrapolation by the use of mathematical models be applied to setting exposure limits. Exposure limits for Class 1 residual solvents could be determined with the use of a large safety factor (i.e., 10,000 to 100,000) with respect to the no-observed-effect level (NOEL). Detection and quantification of these residual solvents should be performed by state-of-the-art analytical techniques.
Acceptable exposure levels in this general chapter for Class 2 residual solvents were established by calculation of PDE values according to the procedures for setting exposure limits in pharmaceuticals (page 5748 of PF 15(6) [Nov.–Dec. 1989]), and the method adopted by IPCS for Assessing Human Health Risk of Chemicals (Environmental Health Criteria 170, WHO, 1994). These procedures are similar to those used by the U.S. EPA (IRIS) and the U.S. FDA (Red Book) and others. The method is outlined here to give a better understanding of the origin of the PDE values. It is not necessary to perform these calculations in order to use the PDE values presented in Table 2 of this document.
PDE is derived from the no-observed-effect level (NOEL), or the lowest-observed effect level (LOEL), in the most relevant animal study as follows:
PDE = (NOEL × Weight Adjustment)/(F1 × F2 × F3 × F4 × F5) (1)
(1)
The PDE is derived preferably from a NOEL. If no NOEL is obtained, the LOEL may be used. Modifying factors proposed here, for relating the data to humans, are the same kind of “uncertainty factors” used in Environmental Health Criteria (Environmental Health Criteria 170, WHO, Geneva, 1994) and “modifying factors” or “safety factors” in Pharmacopeial Forum. The assumption of 100 percent systemic exposure is used in all calculations regardless of route of administration.
The modifying factors are as follows:
| F1 = | A factor to account for extrapolation between species | |
| F1 = | 2 for extrapolation from dogs to humans | |
| F1 = | 2.5 for extrapolation from rabbits to humans | |
| F1 = | 3 for extrapolation from monkeys to humans | |
| F1 = | 5 for extrapolation from rats to humans | |
| F1 = | 10 for extrapolation from other animals to humans | |
| F1 = | 12 for extrapolation from mice to humans | |
F1 takes into account the comparative surface area to body weight ratios for the species concerned and for man. Surface area (S) is calculated as:
S = kM0.67(2)
in which M = body weight, and the constant k has been taken to be 10. The body weights used in the equation are those shown below in Table A3.-1.
| F2 = | A factor of 10 to account for variability between individuals. A factor of 10 is generally given for all organic solvents, and 10 is used consistently in this general chapter. |
| F3 = | A variable factor to account for toxicity studies of short-term exposure | |
| F3 = | 1 for studies that last at least one half-lifetime (1 year for rodents or rabbits; 7 years for cats, dogs, and monkeys) | |
| F3 = | 1 for reproductive studies in which the whole period of organogenesis is covered | |
| F3 = | 2 for a 6-month study in rodents, or a 3.5-year study in nonrodents | |
| F3 = | 5 for a 3-month study in rodents, or a 2-year study in nonrodents | |
| F3 = | 10 for studies of a shorter duration | |
In all cases, the higher factor has been used for study durations between the time points (e.g., a factor of 2 for a 9-month rodent study).
| F4 = | A factor that may be applied in cases of severe toxicity, e.g., nongenotoxic carcinogenicity, neurotoxicity, or teratogenicity. In studies of reproductive toxicity, the following factors are used: | |
| F4 = | 1 for fetal toxicity associated with maternal toxicity |
|
| F4 = | 5 for fetal toxicity without maternal toxicity | |
| F4 = | 5 for a teratogenic effect with maternal toxicity | |
| F4 = | 10 for a teratogenic effect without maternal toxicity |
|
| F5 = | A variable factor that may be applied if the no-effect level was not established |
When only a LOEL is available, a factor of up to 10 can be used, depending on the severity of the toxicity. The weight adjustment assumes an arbitrary adult human body weight for either sex of 50 kilograms (kg). This relatively low weight provides an additional safety factor against the standard weights of 60 kg or 70 kg that are often used in this type of calculation. It is recognized that some adult patients weigh less than 50 kg; these patients are considered to be accommodated by the built-in safety factors used to determine a PDE. If the solvent was present in a formulation specifically intended for pediatric use, an adjustment for a lower body weight would be appropriate.
As an example of the application of this equation, consider a toxicity study of acetonitrile in mice that is summarized in Pharmeuropa, Vol. 9, No. 1, Supplement, April 1997, page S24. The NOEL is calculated to be 50.7 mg kg–1 day–1. The PDE for acetonitrile in this study is calculated as follows:
PDE = (50.7 mg kg1 day1 × 50 kg)/(12 × 10 × 5 × 1 × 1) = 4.22 mg day1
In this example,
| F1 = | 12 to account for the extrapolation from mice to humans |
| F2 = | 10 to account for differences between individual humans |
| F3 = | 5 because the duration of the study was only 13 weeks |
| F4 = | 1 because no severe toxicity was encountered |
| F5 = | 1 because the no-effect level was determined |
A3.-1. Values Used in the Calculations in This Document
| Rat body weight | 425 g |
| Pregnant rat body weight | 330 g |
| Mouse body weight | 28 g |
| Pregnant mouse body weight | 30 g |
| Guinea-pig body weight | 500 g |
| Rhesus monkey body weight | 2.5 kg |
| Rabbit body weight (pregnant or not) | 4 kg |
| Beagle dog body weight | 11.5 kg |
| Rat respiratory volume | 290 L/day |
| Mouse respiratory volume | 43 L/day |
| Rabbit respiratory volume | 1440 L/day |
| Guinea-pig respiratory volume | 430 L/day |
| Human respiratory volume | 28,800 L/day |
| Dog respiratory volume | 9000 L/day |
| Monkey respiratory volume | 1150 L/day |
| Mouse water consumption | 5 mL/day |
| Rat water consumption | 30 mL/day |
| Rat food consumption | 30 g/day |
The equation for an ideal gas, PV = nRT, is used to convert concentrations of gases used in inhalation studies from units of ppm to units of mg/L or mg/m3. Consider as an example the rat reproductive toxicity study by inhalation of carbon tetrachloride (molecular weight 153.84) summarized in Pharmeuropa, Vol. 9, No. 1, Supplement, April 1997, page S9.
+'&img=../../images/v35300/c467-e5-usp33.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
The relationship 1000 L = 1 m3 is used to convert to mg/ m3.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Horacio N. Pappa, Ph.D.
Principal Scientific Liaison 1-301-816-8319 |
(GCCA2010) General Chapters - Chemical Analysis |
USP35–NF30 Page 185
Pharmacopeial Forum: Volume No. 37(1)