INTRODUCTION
Raman spectroscopy shares many of the principles that apply to other spectroscopic measurements discussed in Spectrophotometry and Light-Scattering  851
851 . Raman is a vibrational spectroscopic technique and is therefore related to infrared (IR) and near-infrared (NIR) spectroscopy. The Raman effect itself arises as a result of a change in the polarizability of molecular bonds during a given vibrational mode and is measured as inelastically scattered radiation.
. Raman is a vibrational spectroscopic technique and is therefore related to infrared (IR) and near-infrared (NIR) spectroscopy. The Raman effect itself arises as a result of a change in the polarizability of molecular bonds during a given vibrational mode and is measured as inelastically scattered radiation.
A Raman spectrum is generated by exciting the sample of interest to a virtual state with a monochromatic source, typically a laser. Light elastically scattered (no change in wavelength) is known as Rayleigh scatter and is not of interest in Raman spectrometry, except for marking the laser wavelength. However, if the sample relaxes to a vibrational energy level that differs from the initial state, the scattered radiation is shifted in energy. This shift is commensurate with the energy difference between the initial and final vibrational states. This “inelastically scattered” light is referred to as Raman scatter. Only about one in 106–108 photons incident on the sample undergoes Raman scattering. Thus lasers are employed in Raman spectrometers. If the Raman-scattered photon is of lower energy, it is referred to as Stokes scattering. If it is of higher energy, it is referred to as anti-Stokes scattering. In practice, nearly all analytically useful Raman measurements make use of Stokes-shifted Raman scatter.
The appearance of a Raman spectrum is much like an infrared spectrum plotted linearly in absorbance. The intensities, or the number of Raman photons counted, are plotted against the shifted energies. The x-axis is generally labeled “Raman Shift/cm–1” or “Wavenumber/cm–1”. The Raman shift is usually expressed in wavenumber and represents the difference in the absolute wavenumber of the peak and the laser wavenumber. The spectrum is interpreted in the same manner as the corresponding mid-infrared spectrum. The positions of the (Raman shifted) wavenumbers for a given vibrational mode are identical to the wavenumbers of the corresponding bands in an IR absorption spectrum. However, the stronger peaks in a Raman spectrum are often weak in an IR spectrum, and vice versa. Thus the two spectroscopic techniques are often said to be complementary.
Raman spectroscopy is advantageous because quick and accurate measurements can often be made without destroying the sample (solid, semisolid, liquid or, less frequently, gas) and with minimal or no sample preparation. The Raman spectrum contains information on fundamental vibrational modes of the sample that can yield both sample and process understanding. The signal is typically in the visible or NIR range, allowing efficient coupling to fiber optics. This also means that a signal can be obtained from any medium transparent to the laser light; examples are glass, plastics, or samples in aqueous media. In addition, because Raman spectra are ordinarily excited with visible or NIR radiation, standard glass/quartz optics may be used. From an instrumental point of view, modern systems are easy to use, provide fast analysis times (seconds to several minutes), and are reliable. However, the danger of using high-powered lasers must be recognized, especially when their wavelengths are in the NIR and, therefore, not visible to the eye. Fiber-optic probes should be used with caution and with reference to appropriate government regulations regarding lasers and laser classes.
In addition to “normal” Raman spectroscopy, there are several more specialized Raman techniques. These include resonance Raman (RR), surface-enhanced Raman spectroscopy (SERS), Raman optical activity (ROA), coherent anti-Stokes Raman spectroscopy (CARS), Raman gain or loss spectroscopy, and hyper-Raman spectroscopy. These techniques are not widely employed in pharmaceutical laboratories, and are not addressed in this general information chapter.
QUALITATIVE AND QUANTITATIVE RAMAN MEASUREMENTS
There are two general classes of measurements that are commonly performed by Raman spectrometry: qualitative and quantitative.
Qualitative Raman Measurements
Qualitative Raman measurements yield spectral information about the functional groups that are present in a sample. Because the Raman spectrum is specific for a given compound, qualitative Raman measurements can be used as a compendial ID test, as well as for structural elucidation.
Quantitative Raman Measurements
For instruments equipped with a detector that measures optical power (such as Fourier transform [FT]-Raman spectrometers), quantitative Raman measurements utilize the following relationship between signal, S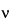 , at a given wavenumber, , and the concentration of an analyte, C:
, at a given wavenumber, , and the concentration of an analyte, C:
S = K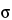 (L
(L 
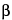 )4P0C
)4P0C
in which K is a constant that depends on laser beam diameter, collection optics, sample volume, and temperature; is the Raman cross section of the particular vibrational mode; L is the laser wavenumber; is the wavenumber of the vibrational mode; and P0 is the laser power. The Raman cross section, V, is characteristic of the nature of the particular vibrational mode. The sample volume is defined by size of the focus of the laser beam at the sample, the optic being used for focusing, and the optical properties of the sample itself. Spot sizes at the sample can range from less than 1 µm for a microprobe to 6 mm for a large area sample system. For Raman spectrometers that measure the number of photons per second (such as change-coupled device [CCD]-Raman spectrometers) the corresponding equation is:
S = KL(L )3P0C
From the above equations, it is apparent that peak signal is directly proportional to concentration. It is this relationship that is the basis for the majority of quantitative Raman applications.
FACTORS AFFECTING QUANTIFICATION
Sample-Based Factors
The most important sample-based factors that deleteriously affect quantitative Raman spectrometry are fluorescence, sample heating, absorption by the matrix or the sample itself, and the effect of polarization. If the sample matrix includes fluorescent compounds, the measured signal will usually contain a contribution from fluorescence. Fluorescence will be observed only if the laser excitation wavelength overlaps with an absorption band of a fluorescent compound. Fluorescence is typically observed as a broad sloping background underlying the Raman spectrum. Fluorescence can cause both a baseline offset and reduced signal-to-noise ratio. The wavelength range and intensity of the fluorescence is dependent on the chemical composition of the fluorescent material. Because fluorescence is generally a much more efficient process than Raman scattering, even very minor amounts of fluorescent impurities can lead to significant degradation of the Raman signal. Fluorescence can be reduced by using longer wavelength excitation sources such as 785 nm or 1064 nm. However, it should be remembered that the strength of the Raman signal is proportional to (L )4, so the advantage of using a long-wavelength excitation laser to minimize fluorescence is at least partially offset by the reduced strength of the Raman signal. The greatest signal-to-noise ratio will be obtained by balancing fluorescence rejection, signal strength, and detector response.
Fluorescence in solids can sometimes be mitigated by exposing the sample to the laser radiation for a period of time before measurement. This process is called photobleaching, and operates by degrading the highly absorbing species. Photobleaching is less effective in liquids, where the sample is mobile, or if the amount of fluorescent material is more than a trace.
Sample heating by the laser source can cause a variety of effects, such as physical form change (melting), polymorph conversion, or sample burning. The chance for sample heating is greatest when the spot size at the sample is the smallest, i.e., when a microprobe is being used. This is usually an issue for colored, highly absorbing species, or very small particles that have low heat transfer. The effects of sample heating are usually observable either as changes in the Raman spectrum over time or by visual inspection of the sample. Besides decreasing the laser flux, a variety of methods can be employed to diminish laser-induced heating, such as moving the sample or laser during the measurement or improving the heat transfer from the sample with thermal contact or liquid immersion.
Absorption of the Raman signal by the matrix or the sample itself can also occur. This problem is more prevalent with long-wavelength FT-Raman systems where the Raman signal can overlap with an NIR overtone absorption. This effect will be dependent on the optics of the system as well as on the sample presentation. Associated with this effect is variability from scattering in solids as a result of packing and particle-size differences. The magnitude of all of these effects, however, is typically less severe than in NIR because of the limited depth of penetration and the relatively narrower wavelength region sampled in Raman spectroscopy.
Finally, it should be recognized that laser radiation is polarized and the Raman spectra of crystalline materials and other oriented samples can differ significantly depending on the way that the sample is mounted. If the Raman spectrometer is capable of producing linearly polarized radiation at the sample then a polarization scrambler is recommended for routine sample analysis.
Sampling Factors
Raman spectroscopy is a zero-background technique, in that the signal at the detector is expected to be zero in the absence of a sample. This situation can be contrasted with absorption spectrometry, where the signal at the detector is at a maximum in the absence of a sample. Zero-background techniques are inherently sensitive because small changes in sample concentration lead to proportionate changes in the signal level. The instrument will also be sensitive to other sources of light that can cause sample-to-sample variations in the measured signal level. In addition, a large background signal caused by fluorescence will lead to an increased noise level (photon shot noise). Thus it may be very difficult to use the absolute Raman signal for direct determination of an analyte. Other potential sources of variation are changes in the sample opacity and heterogeneity, changes in the laser power at the sample, and changes in optical collection geometry or sample position. These effects can be minimized by sampling in a reproducible, representative manner. Careful design of the instrumentation can reduce these effects but they cannot be eliminated entirely.
Use of an internal reference standard is the most common and robust method of eliminating variations caused by absolute intensity fluctuations. There are several choices for this approach. An internal standard can be deliberately added, and isolated peaks from this standard can be employed; or a band due to a moiety such as an aromatic ring, the Raman cross-section of which does not change with the way the sample is prepared, can also be used. For solution spectra, an isolated solvent band can be employed because the solvent will remain relatively unchanged from sample to sample. Also, in a formulation, an excipient peak can be used if it is in substantial excess compared to the analyte. The entire spectrum can also be used as a reference, with the assumption that laser and sample-orientation changes will affect the entire spectrum equally.
A second important sampling-based factor to consider is spectral contamination. Raman scattering is a weak effect that can be masked by a number of external sources. Common contamination sources include sample-holder artifacts (container or substrate) and ambient light. Typically, these issues can be identified and resolved by careful experimentation.
APPARATUS
Components
All modern Raman measurements involve irradiating a sample with a laser, collecting the scattered radiation, rejecting the Rayleigh-scattered light, differentiating the Raman photons by wavelength, and detecting the resulting Raman spectrum. All commercial Raman instruments therefore share the following common features to perform these functions:
- Excitation source (laser)
- Sampling device
- Device to filter/reject light scattered at the laser wavelength
- Wavelength processing unit
- Detector and electronics
excitation source (laser)
Table 1 identifies several common lasers used for pharmaceutical applications or Raman spectrometry. UV lasers have also been used for specialized applications but have various drawbacks that limit their utility for general analytical measurements. As more applications for UV lasers are described, it is likely that they may become more common for Raman spectrometry.
Table 1. Lasers Used in Pharmaceutical Applications
| Laser whole number) |
Type | Typical Power at Laser |
Wavelength Range, nm (Stokes Region, 100 cm–1 to 3000 cm–1 shift) |
Comments |
|---|---|---|---|---|
| NIR Lasers | ||||
| 1064 | Solid state (Nd:YAG) |
Up to 3 W | 1075–1563 | Commonly used in Fourier transform instruments |
| 830 | Diode | Up to 300 mW | 827–980 | Typically limited to 2000 cm |
| 785 | Diode | Up to 500 mW | 791–1027 | Most widely used dispersive Raman laser |
| Visible Lasers | ||||
| 632.8 | He–Ne | Up to 500 mW | 637–781 | Relatively small fluorescence risk |
| 532 | Doubled (Nd:YAG) |
Up to 1 W | 535–632.8 | High fluorescence risk |
| 514.5 | Ar+ | Up to 1 W | 517–608 | High fluorescence risk |
| 488–632.8 | Ar+ | Up to 1 W | 490–572 | High fluorescence risk |
sampling device
Several sampling arrangements are possible, including direct optical interfaces, microscopes, fiber optic-based probes (either noncontact or immersion optics), and sample chambers (including specialty sample holders and automated sample changers). The sampling optics can also be designed to obtain the polarization-dependent Raman spectrum, which often contains additional information. Selection of the sampling device will often be dictated by the analyte and sample. However, considerations such as sampling volume, speed of the measurement, laser safety, and reproducibility of sample presentation should be evaluated to optimize the sampling device for any given application.
filtering device
The intensity of scattered light at the laser wavelength (Rayleigh) is many orders of magnitude greater than the Raman signal and must be rejected prior to the detector. Notch filters are almost universally used for this purpose and provide excellent rejection and stability combined with small size. The traditional use of multistage monochromators for this purpose, although still viable, is now rare. In addition, various filters or physical barriers to shield the sample from external radiation sources (e.g., room lights, laser plasma lines) may be required depending on the collection geometry of the instrument.
wavelength processing unit
The wavelength scale may be encoded by either a scanning monochromator, a grating polychromator (in CCD-Raman spectrometers) or a two-beam interferometer (in FT-Raman spectrometers). A discussion of the specific benefits and drawbacks of each of the dispersive designs compared to the FT instrument is beyond the scope of this chapter. Any properly qualified instruments should be suitable for qualitative measurements. However, care must be taken when selecting an instrument for quantitative measurements, as dispersion and response linearity might not be uniform across the full spectral range.
detector
The silicon-based CCD array is the most common detector for dispersive instruments. The cooled array detector allows measurements over the spectral range from 4500 to 100 cm1 Raman shift with low noise when most visible lasers, such as frequency-doubled neodymium-doped yttrium–aluminum–garnet (Nd:YAG) (532 nm) or helium–neon (632.8 nm) lasers, are used. When a 785-nm diode laser is used, the wavelength range is reduced to about 3100 to 100 cm1. The most commonly used CCD has its peak wavelength responsivity when matched to the commonly used 632.8-nm He–Ne gas laser or 785-nm diode laser. FT instruments typically use single-channel germanium or indium–gallium–arsenide (InGaAs) detectors responsive in the NIR to match the 1064-nm excitation of a Nd:YAG laser.
Calibration
Raman instrument calibration involves three components: primary wavelength (x-axis), laser wavelength, and intensity (y-axis).
primary wavelength (x-axis)
In the case of FT-Raman instruments, primary wavelength-axis calibration is maintained, at least to a first approximation, with an internal He–Ne laser. Most dispersive instruments utilize atomic emission lamps for primary wavelength-axis calibration. In all instruments suitable for analytical Raman measurements, the vendor will offer a procedure of x-axis calibration that can be performed by the user. For dispersive Raman instruments, a calibration based on multiple atomic emission lines is preferred. The validity of this calibration approach can be verified subsequent to laser wavelength calibration by using a suitable Raman shift standard. For scanning dispersive instruments, calibration might need to be performed more frequently, and precision in both a scanning and static operation mode may need to be verified.1
laser wavelength
Laser wavelength variation can impact both the wavelength precision and the photometric (signal) precision of a given instrument. Even the most stable current lasers can vary slightly in their measured wavelength output. The laser wavelength must therefore be confirmed to ensure that the Raman shift positions are accurate for both FT-Raman or dispersive Raman instruments. A reference Raman shift standard material such as those outlined in ASTM E1840-96 (2002)1 or other suitably verified materials can be utilized for this purpose. [Note—Reliable Raman shift standard values for frequently used liquid and solid reagents, required for wavenumber calibration of Raman spectrometers, are provided in the ASTM Standard Guide cited. These values can be used in addition to the highly accurate and precise low-pressure arc lamp emission lines that are also available for use in Raman instrument calibration. ] Spectrometric grade material can be purchased from appropriate suppliers for this use. Certain instruments may use an internal Raman standard separate from the primary optical path. External calibration devices exactly reproduce the optical path taken by the scattered radiation. [Note—When chemical standards are used, care must be taken to avoid contamination and to confirm standard stability. ]
Unless the instrument is of a continuous calibration type, the primary wavelength axis calibration should be performed, as per vendor procedures, just prior to measuring the laser wavelength. For external calibration, the Raman shift standard should be placed at the sample location and measured using appropriate acquisition parameters. The peak center of a strong, well-resolved band in the spectral region of interest should be evaluated. The position can be assessed manually or with a suitable, valid peak-picking algorithm. The software provided by the vendor might measure the laser wavelength and adjust the laser wavelength appropriately so that this peak is at the proper position. If the vendor does not provide this functionality, the laser wavelength should be adjusted manually. Depending on the type of laser, the laser wavelength can vary with temperature, current, and voltage. Wavelength tolerances can vary depending on the specific application.
signal level (y-axis)
Calibration of the photometric axis can be critical for successful quantification by using certain analytical methods (chemometrics) and method transfer between instruments. Both FT-Raman and dispersive Raman spectrometers should undergo similar calibration procedures. The tolerance of photometric precision acceptable for a given measurement should be assessed during the method development stage.
To calibrate the photometric response of a Raman instrument, a broad-band emission source should be used. There are two accepted methods. Method A utilizes a tungsten white light source.2 The output power of such sources is traceable to the National Metrology Institute (NMI). In the United Kingdom, the National Physical Laboratory also provides calibrated light bulbs. Several other vendors also provide NIST-traceable irradiance calibration standards. This method is applicable to all common laser excitation wavelengths listed in Table 1. In Method B, NIST standard reference materials (SRMs) are utilized.3 Several doped-glass fluorescence standards are currently available.
Method A—
The source should be placed at the sample location with the laser off and the response of the detector measured (using parameters appropriate for the instrument). The output for the source used for calibration should be known. The ratio of the measured response to the true response should be determined and a correction file generated. This correction should be applied to all spectra acquired with the instrument. Most manufacturers will provide both appropriate calibration sources and software for this approach. If the manufacturer does not provide a procedure or method, the user can accomplish the task using a source obtained from NIST and appropriate software. If a manufacturer's method is used, attention must be paid to the calibration procedure and source validity. The user should obtain appropriate documentation from the manufacturer to ensure a qualified approach.
Method B—
The fluorescence standard should be placed at the sample location. With the laser on, a spectrum of the SRM should be obtained (using parameters appropriate for the instrument). The output of the source used for calibration should be known. The ratio of the measured response to the true response should be determined and a correction file generated. This correction should be applied to all spectra acquired with the instrument. Most manufacturers will provide both appropriate calibration sources and software for this approach. If the manufacturer does not provide a procedure or method, the user can accomplish the task using a source obtained from NIST and appropriate software. If a manufacturer's method is used, attention must be paid to the calibration procedure and source validity. The user should obtain appropriate documentation from the manufacturer to ensure a qualified approach. [Note—Method B is currently appropriate for systems with 785-nm (SRM 2241), 532-nm (SRM 2242), and both 514.5-nm and 488-nm (SRM 2243) laser excitation. NIST is currently developing other SRMs that will be wavelength-specific for 1064-nm (SRM 2244) and 632.8-nm excitation (expected to be available in 2006). ]
external calibration
Detailed functional validation employing external reference standards is recommended to demonstrate instrumental suitability for laboratory instruments, even for instruments that possess an internal calibration approach. The use of external reference standards does not obviate the need for internal quality control procedures; rather, it provides independent documentation of the fitness of the instrument to perform the specific analysis or purpose. For instruments installed in a process location or in a reactor where positioning of an external standard routinely is not possible, including those instruments that employ an internal calibration approach, the relative performance of an internal versus an external calibration approach should be periodically checked. The purpose of this test is to check for changes in components that might not be included in the internal calibration method (process lens, fiber-optic probe, etc.), e.g., photometric calibration of the optical system.
QUALIFICATION AND VERIFICATION OF RAMAN SPECTROMETERS
The suitability of a specific instrument for a given method is ensured by a thorough technology-suitability evaluation for the application; a routine, periodic instrument operational qualification; and the more frequent performance verification (see Definition of Terms and Symbols). The purpose of the technology-suitability evaluation is to ensure that the technology proposed is suitable for the intended application. The purpose of the instrument qualification is to ensure that the instrument to be used is suitable for its intended application and, when requalified periodically, continues to function properly over extended time periods. When the device is used for a specific qualitative or quantitative analysis, regular performance verifications are made. Because there are many different approaches to measuring Raman spectra, instrument operational qualification and performance verification often employ external standards that can be used on any instrument. As with any spectrometric device, a Raman instrument needs to be qualified for both wavenumber (x-axis and shift from the excitation source) and photometric (signal axis) precision.
In performance verification, a quality-of-fit to an initial scan or group of scans (often referred to in nonscanning instruments as an accumulation) included in the instrumental qualification can be employed. In such an analysis, it is assumed that reference standard spectra collected on a new or a newly repaired, properly operating instrument represent the best available spectra. Comparison of spectra taken over time on identical reference standards (either the original standard or identical new standards, if stability of the reference standards is a concern) forms the basis for evaluating the long-term stability of a Raman measurement system.
Frequency of Testing
Instrumental qualification is performed at designated intervals or following a repair or significant optical reconfiguration, such as the replacement of the laser, the detector or the notch or edge filters. Full instrument requalification might not be necessary when changing between sampling accessories such as a microprobe, a sample compartment, or a fixed fiber-optic probe. Performance verification tests may be sufficient in these cases; instrument-specific guidance from the vendor on qualification requirements should be followed. Tests include wavelength (x-axis and shift from the excitation source) and photometric (signal axis) precision. Instrument qualification tests require that specific application-dependent tolerances be met.
Performance verification is carried out on the instrument configured for the analytical measurements and is performed more frequently than instrument qualification. Performance verification includes measurement of the wavelength uncertainty and intensity-scale precision. Wavelength precision and intensity-scale precision tests may be needed prior to any data collection on a given day. Performance is verified by matching the current spectra to those collected during the previous instrument qualification.
Instrument Operational Qualification
It is important to note that the acceptance specifications given in both the Instrument Operational Qualification and Performance Qualification sections are applicable for general use; specifications for particular instruments and applications can vary depending on the analysis method used and the desired accuracy of the final result. ASTM standard reference materials are also specified, with the understanding that under some circumstances (specifically remote on-line applications) calibration using one of these materials may be impractical, and other suitably verified materials can be employed. At this juncture it is important to note that specific parameters such as spectrometer noise, limits of detection (LOD), limits of quantification (LOQ), and acceptable spectral bandwidth for any given application should be included as part of the analytical method development. Specific values for tests such as spectrometer noise and bandwidth will be dependent on the instrument chosen and the purpose required. As a result, specific instrument tests for these parameters are not dictated in this information chapter.
wavelength (x-axis) accuracy
It is important to ensure the accuracy of the wavelength axis via calibration to maintain the integrity of Raman peak positions. Wavelength calibration of a Raman spectrometer consists of two parts: primary wavelength axis and laser wavelength calibration. After both the primary wavelength axis and the laser wavelength are calibrated, instrument wavelength uncertainty can be determined. This can be accomplished using a Raman shift standard such as the ASTM shift standards or other suitably verified material. Selection of a standard with bands present across the full Raman spectral range is recommended so that instrument wavelength uncertainty can be evaluated at multiple locations within the spectrum. The tolerance of wavelength precision that is required for a given measurement should be assessed during the method-development stage. [Note—For scanning dispersive instruments, calibration might need to be performed more frequently, and precision in both a scanning and static operation mode may need to be verified. ]
photometric precision
Laser variation in terms of the total emitted photons occurring between two measurements can give rise to changes in the photometric precision of the instrument. Unfortunately, it is very difficult to separate changes in the photometric response associated with variations in the total emitted laser photons from the sample- and sampling-induced perturbations. This is one of the reasons why absolute Raman measurements are strongly discouraged and why the photometric precision specification is set relatively loosely. The tolerance of photometric precision required for a given measurement should be assessed during the method-development stage.
performance qualification
The objective of performance qualification is to ensure that the instrument is performing within specified limits with respect to wavelength precision, photometric axis precision, and sensitivity. In certain cases when the instrument has been set up for a specific measurement (for example, installed in a process reactor), it might no longer be possible or desirable to measure the wavelength and photometric (signal) qualification reference standards identified above. Provided instrument operational qualification has shown that the equipment is fit for use, a single external performance verification standard can be used to reverify function on a continuing basis (for example, a routinely used process solvent signal, for both wavelength and photometric precision, following reactor cleaning). The performance verification standard should match the format of the samples in the current analysis as closely as possible and use similar spectral acquisition parameters. Quantitative measurements of an external performance verification standard spectrum check both the wavelength (x-axis and laser wavelength) and the photometric (signal) precision. Favorable comparison of a series of performance verification spectra demonstrates proper continued operation of the instrument.
wavelength precision
The wavelength precision should be measured by collecting data for a single spectrum of the selected Raman shift standard for a period equal to that used in the photometric consistency test. When appropriate, powdered samples should be repacked between each set of measurements. Peak positions across the spectral range of interest are used to calculate precision. Performance is verified by matching the current peak positions to those collected during the previous instrument qualification and should not vary with a standard deviation of more than ±0.3 cm–1, although this specification can be adjusted according to the required accuracy of the measurement.
photometric precision
The photometric precision should be measured by collecting data for a single spectrum of a suitably verified reference standard material for a specified time. After suitable baseline correction, the areas of a number of bands across the spectral range of interest should be calculated by means of an appropriate algorithm. The area of the strongest band is set to 1, and all other envelopes are normalized to this band. Performance is verified by matching the current band areas to the respective areas collected during the previous instrument qualification. The areas should vary by no more than 10%, although this specification can be adjusted according to the required accuracy of the measurement.
laser power output precision and accuracy
This test is applicable only to Raman instruments with automatic, internal laser power meters. Instruments without laser power measurement should utilize a calibrated laser power meter from a reputable supplier. The laser output should be set to a representative output, dictated by the requirements of the analytical measurement and the laser power measured. The output should be measured and checked against the output measured at instrument qualification. The power (in milliwatts or watts) should vary by no more than 25% compared to the qualified level. If the power varies by more than this amount, the instrument should be serviced (as this variation might indicate, among other things, a gross misalignment of the system or the onset of failure of the laser).
For instruments with an automatic, internal laser power meter, the accuracy of the values generated from the internal power meter should be compared to a calibrated external laser power meter at an interval of not more than 12 months. The internally calculated value should be compared to that generated by the external power meter. Performance is verified by matching the current value to that generated during the previous instrument qualification. The manufacturer might provide software to facilitate this analysis. If the instrument design prevents the use of an external power meter, then the supplier should produce documentation to ensure the quality of the instrument and provide a recommended procedure for the above analysis to be accomplished during a scheduled service visit.
METHOD VALIDATION
Validation of Raman methods will follow the same protocols described in Validation of Compendial Procedures 1225 in terms of accuracy, precision, etc. However, several of these criteria are affected by variables specific to Raman spectrometry. Fluorescence is the primary variable that can affect the suitability of a method. The presence of fluorescent impurities in samples can be quite variable and have little effect on the acceptability of a material. The method must be flexible enough to accommodate different sampling regimes that may be necessary to minimize the effects of these impurities.
Detector linearity must be confirmed over the range of possible signal levels. Fluorescence might drive both the signal baseline and the noise higher than that used in the validation, in which case the fluorescence must be decreased, or the method modified to accommodate the higher fluorescence levels. This is also true for the precision, limit of detection, and limit of quantification of the method, as increased baseline noise will negatively impact all of these values. Because fluorescence can also affect quantification caused by baseline shifts, acceptable quantification at different levels of photobleaching, when used, should also be confirmed.
The impact of the laser on the sample must be determined. Visual inspection of the sample and qualitative inspection of the Raman spectrum for measurements with differing laser powers and exposure times will confirm that the sample is not being altered (other than by photobleaching). Specific variables to confirm in the spectrum are shifts in peak position, changes in peak height and band width, and unexpected changes in background intensity.
Method precision must also encompass sample position. The sample presentation is a critical factor for both solids and liquids, and must be either tightly controlled or accounted for in the calibration model. Sample-position sensitivity can often be minimized by appropriate sample preparation or sample holder geometry, but will vary from instrument to instrument based on excitation and collection optical configuration.
DEFINITION OF TERMS AND SYMBOLS
calibration model is a mathematical expression that relates the response from an analytical instrument to the properties of samples.
instrument bandpass (or resolution) is a measure of the capability of a spectrometer to separate radiation of similar wavelengths.
operational qualification is the process by which it is demonstrated and documented that the instrument performs according to specifications, and that it can perform the intended task. This process is required following any significant change such as instrument installation, relocation, major repair, etc.
performance qualification is the process of using one or more well-characterized and stable reference materials to verify consistent instrument performance. Qualification may employ the same or different standards for different performance characteristics.
raman spectra4 are plots of the radiant energy, or number of photons, scattered by the sample through the indirect interaction between the molecular vibrations in the sample and monochromatic radiation of frequency much higher than that of the vibrations. The abscissa is usually the difference in wavenumber between the incident and scattered radiation.
(normal) raman scattering4 is the inelastic scattering of radiation that occurs because of changes in the polarizability, of the relevant bonds during a molecular vibration. Normal Raman spectra are excited by radiation that is not in resonance with electronic transitions in the sample.
raman wavenumber shift4,
+'&img=../../images/v35300/c1120-f2-pf324.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
is the wavenumber of the exciting line minus the wavenumber of the scattered radiation. SI unit: m1. Common unit: cm1 = 100 m1.
+'&img=../../images/v35300/c1120-f1-pf324.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
where is the differential Raman cross section, is positive for Stokes scattering and negative for anti-Stokes scattering.
1
ASTM E1840-96 (2002) Standard Guide for Raman Shift Standards for Spectrometer Calibration, ASTM International, 100 Barr Harbor Drive, PO Box C700, West Conshohocken, PA, USA 19428-2959.
2
NIST-traceable tungsten white light source statement: While the calibration of the Raman frequency (or Raman shift, cm–1) axis using pure materials and an existing ASTM standard is well accepted, techniques for calibration of the Raman intensity axis are not. Intensity calibrations of Raman spectra can be accomplished with certified white light sources.
3
NIST SRM 2241: Ray KG, McCreery RL. Raman intensity correction standard for systems operating with 785-nm excitation. Appl. Spectrosc. 1997, 51, 108–116.
4
Chalmers, J., Griffiths, P., Eds. Handbook of Vibrational Spectroscopy; John Wiley & Sons, Ltd: New York, 2002.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Horacio N. Pappa, Ph.D.
Principal Scientific Liaison 1-301-816-8319 |
(GCCA2010) General Chapters - Chemical Analysis |
USP35–NF30 Page 720
Pharmacopeial Forum: Volume No. 32(4) Page 1211