SCOPE
This chapter provides a consolidated source of information regarding human plasma, with emphasis on plasma for fractionation. Specifically, the chapter addresses plasma classification and nomenclature; collection and processing procedures required for ensuring product safety; details of specific plasmas; and quality systems relating to plasma collection. The chapter also includes, at the end of the text sections, a glossary; a list of abbreviations used in the chapter; and appendices that provide plasma definitions, donor selection criteria, and testing requirements.
Plasma originating from U.S.-licensed collection facilities provides the major supply for the global plasma derivative market. The U.S. Food and Drug Administration (FDA) regulates the collection and processing of plasma used for further manufacture. Title 21 of the Code of Federal Regulations (CFR) details Good Manufacturing Practice (GMP) requirements and product standards to protect the health of the blood donor and to ensure the safety and efficacy of blood products. CFR regulations are updated periodically, but between revisions, existing regulations may not address the most current issues and scientific developments. Therefore, the FDA periodically publishes guidance documents.1 (For further information, see Quality Systems, below).
This chapter emphasizes U.S. practices, but because plasma and its derivatives are shipped globally, it is important to recognize that there are regional differences in recommendations, requirements, and regulations. Therefore this chapter provides information regarding the European Union (EU), the United Kingdom (UK), and Australia.
OVERVIEW
Composition of Plasma
Plasma constitutes approximately 55% of the total blood volume. It is a clear, straw-colored, complex liquid that is 7% protein, 91% water, and 0.9% mineral salts. The majority (approximately 70%) of total plasma protein is albumin. Additional plasma proteins relevant to fractionation include immunoglobulins, coagulation factors, fibrinolytic proteins, proteases, and protease inhibitors. These constituent plasma proteins can be isolated on the basis of the different solubility characteristics of each protein when subjected to specific conditions of pH, temperature, ionic strength, and ethanol concentration. The major products derived from fractionation are listed in Table 1.
Table 1. The Major Fractions and Products from the Cohn Process
| Fraction | Product |
|---|---|
| Cryoprecipitate | Antihemophilic factor (FVIII) |
| Cryosupernatant | Antithrombin III, factor IX complex |
| Fraction I | Fibrinogen, factor XIII |
| Fraction II | Immune globulin G (IgG) |
| Fraction III | IgA, IgM, prothrombin, plasminogen |
| Fraction IV-1 | Factor IX complex, activated factor IX complex |
| Fraction IV-4 | Plasma protein fraction, alpha-1 proteinase inhibitor |
| Fraction V | Albumin |
Plasma for Manufacture of Derivative Products
The two methods for collection of human plasma are automated apheresis (for definitions, see the Glossary preceding the appendices) and centrifugation of whole blood donations. Source Plasma collected by apheresis constitutes the majority of plasma used in the manufacture of plasma derivatives in the United States. Plasma collected for transfusion but not so used (i.e., recovered plasma) also may be used for manufacture. Flow charts delineating how apheresis and whole blood–derived plasma can be used in the manufacture of plasma derivatives in the United States and Europe are presented in Figures 1 and 2, respectively.
+'&img=../../images/v35300/m2497-f1-pf352.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 1. U.S. plasma derivative manufacture: FDA standards.
+'&img=../../images/v35300/m2497-f2-pf352.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 2. EU plasma derivative manufacture: EU standards.
Regardless of the collection method, plasma for fractionation should be a clear to slightly turbid liquid without visible sign of hemolysis; it may vary in color from light yellow to green; it should be ±10% of the stated volume; and it should show no sign of clots.
Source Plasma
Licensed Source Plasma may be manufactured only in collection centers that are approved by the FDA for the collection and distribution of Source Plasma in interstate commerce. Currently, federal regulations governing the manufacture of Source Plasma, including minimal requirements for donors, are found in 21 CFR 640, Subpart G. By definition, Source Plasma is plasma intended for further manufacture. Source Plasma donors can donate as often as twice a week and may be compensated. In addition to FDA requirements, most plasma collectors and fractionators also comply with voluntary standards established by the Plasma Protein Therapeutics Association (PPTA), a trade and standards-setting organization.2 PPTA voluntary standards address several areas of donor, plasma unit, plasma pool, and center management, and are designed to supplement existing regulatory requirements.
Plasma for Transfusion
Plasma for transfusion is not intended for further manufacture but for direct transfusion to patients. It may be collected by either whole blood or apheresis donation. In the United States, plasma for transfusion comes from unpaid volunteer donors. Blood collecting facilities that collect plasma for transfusion typically comply with requirements of both the CFR and a voluntary trade organization, the American Association of Blood Banks (AABB). Currently, sections 640.3 and 640.31 of 21 CFR outline requirements for donors of whole blood and therefore govern most donors of plasma for transfusion, regardless of the collection method. AABB voluntary standards include information contained in FDA regulations and guidance pertaining to blood and plasma, as well as additional standards.3 Plasma for transfusion may be converted to recovered plasma, an unlicensed product that may be used for further manufacture.
Plasma for Ancillary Use in Biologics Manufacturing
Human plasma and its derivatives are used in the manufacture of other biologic products. In this role, the plasma or plasma derivative falls into the category of ancillary use. This is defined as use of a reagent or material as a processing or purification aid or a reagent that exerts an effect on the therapeutic substance but is not intended to be part of the final product formulation (see the USP general information chapter Ancillary Materials for Cell, Gene, and Tissue-Engineered Products  1043
1043 ).
).
Human plasma is commonly used in manufacturing processes that involve primary human cells or cell lines intended for therapeutic applications. In these applications, plasma provides a source of protein and possibly other factors that enhance expansion and differentiation of cell populations. A variety of methods have been used to prepare human plasma and derivatives for ancillary use, but the practices are not standardized. Allogeneic plasma typically is obtained from either apheresis or whole blood, using citrate anticoagulation. Allogeneic plasma typically is collected from paid donors who, like Source Plasma donors, have been screened for the absence of transfusion-transmissible diseases. Preferred donors may be blood type group AB, because they lack anti-A and anti-B isohemagglutinins. Other preferred donors include untransfused males, because this group is unlikely to have human leukocyte antigen (HLA) antibodies that could react with cells in a given culture system. Serum is prepared either from nonanticoagulated whole blood that has been allowed to clot or by the addition of calcium to plasma obtained from citrated whole blood or apheresis. Heating plasma or serum to 56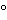 inactivates heat-labile complement and other proteins.
inactivates heat-labile complement and other proteins.
Although there are no standardized specifications for plasma products used as ancillary materials, assays often include safety testing associated with general biologics (e.g., bacterial/fungal cultures, endotoxin, and mycoplasma). In addition, characterization of the products may include tests for irregular erythrocyte and HLA antibodies, osmolality, pH, total protein and immunoglobulin concentrations, hemoglobin concentration, and chemistries such as Na, K, Cl, Ca, and glucose.
In some instances, plasma from bovine sources has been used instead of human plasma. The differences between bovine and human plasma products include factors relevant to their efficacy as ancillary materials, as well as safety of the final manufactured product when administered to humans (see the USP general information chapter Bovine Serum 1024). Fetal bovine serum is preferred to human serum for some applications because it may be superior in promoting human cell growth in vitro. Safety considerations, especially the risk of transmissible spongiform encephalopathy and allergic reactions related to antibovine antibodies, have led to recommendations that human plasma sources be used whenever possible instead of bovine sources. However, substitution of human for bovine plasma does not completely eliminate the risk of infectious and immunologic sequelae, even when human plasma is used as an ancillary material.
PLASMA COLLECTION AND PROCESSING
This section discusses the standard principles involved in plasma collection and the methods used to ensure the safety of the plasma and subsequently manufactured plasma derivatives. Principles for screening and testing of donors are presented in other parts of this chapter.
Collection
Source Plasma is collected by apheresis. Recovered plasma can be obtained either by apheresis or as a by-product of whole blood collection. Collection should take place via an FDA-approved, closed, sterile, pyrogen-free collection system that contains an anticoagulant. No antibacterial or antifungal agent should be added to the plasma. Donations must be collected aseptically. The skin of the donor must be aseptically prepared. Source Plasma is collected using 4% sodium citrate as the anticoagulant. The composition of sodium citrate is given in Table 2.
Three anticoagulant solutions are licensed in the United States for collection of whole blood: citrate phosphate dextrose (CPD), citrate phosphate double dextrose (CP2D), and citrate phosphate dextrose adenine (CPDA-1). The composition of blood collection bags containing these anticoagulants is shown in Table 3. Plasma for transfusion or further manufacture can be made from a unit of whole blood collected in any of the three anticoagulant solutions. Regulations relating to plasma make no distinction among the three anticoagulant solutions. Consequently, collection, storage, and transport requirements are identical regardless of the anticoagulant solution used in the primary collection.
Table 2. Anticoagulant Solution for Collection of Source Plasma by Apheresis (4% Sodium Citrate)
| Volume | Sodium Citrate
Dihydrate |
Citric Acid Anhydrous | pH (25 |
Ratio of Solution to
Whole Blood |
|---|---|---|---|---|
| 250 mL or 500 mL | 40 g/L | As required for pH adjustment | 6.4–7.5 | 1:16 |
Table 3. Anticoagulant Solutions Used during Whole Blood Collection for Recovery of Plasma (500-mL collection bags)
| Anticoagulant | CPDa | CP2Db | CPDA-1c |
| Volume (mL) | 70 | 63 | 63 |
| Dextrose (mg) | 1780 | 3220 | 2010 |
| Sodium citrate dihydrate (mg) | 1840 | 1660 | 1660 |
| Citric acid anhydrous (mg) | 209 | 206 | 206 |
| Monobasic sodium phosphate (mg) | 155 | 140 | 140 |
| Adenine (mg) | — | — | 17.3 |
| pH (25 |
5.3–5.9 | 5.3–5.9 | 5.3–5.9 |
| Ratio of solution to whole blood | 1.4:10 | 1.4:10 | 1.4:10 |
|
Note: Collection of Source Plasma typically involves the use of sodium citrate as the anticoagulant. The specification for sodium citrate is given in Table 2. Plasma for transfusion is stored at 2
a
Citrate phosphate dextrose.
b
Citrate phosphate double dextrose.
c
Citrate phosphate dextrose adenine.
|
|||
Labeling
The labeling for Source Plasma should comply with 21 CFR 640.70 and 21 CFR 640.69(b). The labeling for whole blood should comply with 21 CFR 606.121 and 606.122, and with internal licenses.
A unique identification number is assigned so that the donation can be related to the individual donor records and test results. The origin of each donation in a plasma pool and the results of the corresponding donation and laboratory tests must be traceable while the required degree of confidentiality concerning the donor's identity is maintained. Whole blood must be labeled “This Product may transmit infectious agents” [21 CFR 121(c)(9)]. Source Plasma or recovered plasma must be labeled “Caution: For Manufacturing Use Only” if the product is intended for use in fractionation. For plasma to be used as a reagent or for in vitro use, the required labeling statement is “Caution: For Use in Manufacturing Noninjectable Products Only” [21 CFR 121(e)(5)(ii)].
Storage
Plasma for fractionation should be stored at or below 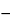 20. The plasma can still be used for fractionation if its temperature exceeds 20 on (at most) one occasion for not more than 72 hours and if the plasma has been maintained at a temperature of 5 or lower at all times. Storage temperatures must be maintained during transport.
20. The plasma can still be used for fractionation if its temperature exceeds 20 on (at most) one occasion for not more than 72 hours and if the plasma has been maintained at a temperature of 5 or lower at all times. Storage temperatures must be maintained during transport.
PLASMA SAFETY CONSIDERATIONS
Plasma is protected by five overlapping safeguards that the FDA has termed the “Five-Layer Safety Net”: donor screening, blood testing, donor deferral, quarantine, and investigation. For guidance in this area, see FDA Publication No. FS 02-1 February 2002.
Voluntary measures that provide an additional margin of safety include recruitment and retention of suitable donors and inventory hold procedures. In the manufacture of plasma-derived products, steps taken for viral clearance are very important for ensuring safety.
None of these measures is sufficient by itself; the safety net is the overlapping combination of the activities.
Donor Screening
The selection of a suitable site for blood and plasma donation activities is a first and very important step to ensure safe donations. Areas with low disease prevalence are preferred as locations for donation centers, thereby reducing the likelihood of collecting plasma from an infected donor.
One of the PPTA voluntary standards is the viral marker standard, which obliges plasma centers to report viral marker rates for human immunodeficiency virus (HIV), hepatitis B virus (HBV), and hepatitis C virus (HCV) in the donor populations. A center's rates are compared to the industry average. Alert limits are set to take into account the number of annual donations in the center. If a center exceeds the limit for any of these viruses or the aggregate of all three, the center must implement corrective actions that will bring the center into compliance with the standard.
Appropriate donor selection helps provide a safe plasma supply. A detailed donor history questionnaire in conjunction with a careful medical examination allows center personnel to recognize unsuitable donors whose behavior puts them at risk for transfusion-transmitted disease or who have underlying medical conditions that preclude donation.
An additional measure put in place by centers that collect Source Plasma is the PPTA National Donor Deferral Registry (NDDR). It lists donors throughout the United States who have been previously deferred from donation (although it provides no information about the reason for deferral). Other countries have different systems depending on their national regulations concerning personal data gathering. Any individual who tests positive for HIV, HBV, or HCV is entered into the national database (the National Donor Deferral Registry) used by all U.S. plasma centers that are certified under the International Quality Plasma Program (IQPP). All individuals who present at a U.S. plasma center for the first time are checked against the NDDR. In this manner, donors who have previously been deferred for positive test results at any participating facility can be identified and rejected quickly. This standard ensures that donors deferred for positive test results do not donate in other facilities.
A voluntary safety initiative, the Qualified Donor Standard, implemented by the plasma fractionation industry, builds on the fact that many plasmapheresis donors contribute plasma frequently. A donor who enters a plasmapheresis center for the first time is called an Applicant Donor, and the first donation is used for further manufacturing only if the donor returns a second time. Potential donors must pass two separate medical screenings and testing for HIV, HBV, and HCV on two different occasions. Only after satisfactory screenings and negative test results does that person become a Qualified Donor. If a donor does not return within 6 months, that person loses his/her Qualified Donor status and must qualify again. This standard means that plasma from a one-time-only donor (even when all test results are negative) cannot be used for further manufacture. This standard results in committed donors and eliminates the risk that plasma centers will accept so-called test seekers. The interval between permitted donations of whole blood is too long to allow a similar screening program, although quite a number of donors in whole-blood donor centers are regular and repetitive donors.
Another PPTA voluntary standard addresses donor management criteria. The Community-Based Donor Standard allows only donors who permanently reside within its defined donor recruitment area to donate at a given center. In addition, a Donor Education Standard requires new donors to engage in an educational program and follow-up assessment regarding HIV/acquired immune deficiency syndrome (AIDS) and activities that place them at risk for HIV/AIDS.
In addition to donor management strategies and standards, PPTA has issued a plasma unit management standard called Inventory Hold. This standard states that collected plasma will be held in inventory for at least 60 days from the time of collection. This allows the retrieval of units as a result of post-donation information (information that was not known at the time of donation) that would have disqualified the donor. This information could include admitting high-risk behavior; becoming reactive for HIV, HBV, or HCV; or providing incorrect information about international travel.
Blood Testing
Testing of donations is an important safety measure both for plasma intended for transfusion and plasma intended for further manufacturing. Both enzyme-linked immunosorbent assays (ELISA) and nucleic acid amplification technologies (NAT) are used to screen donations for the presence of infectious disease. Automation provides the necessary throughput to screen every donation for a variety of potential pathogens.
Testing strategies differ depending on whether the plasma donation is intended for transfusion or further manufacture. Plasma for transfusion requires more extensive infectious disease testing, because there are no pathogen inactivation/removal technologies licensed for this product in the United States. On the other hand, plasma for further manufacture is subjected to several pathogen inactivation/removal steps during manufacture, thereby obviating the need for some disease testing. Table 4 outlines current infectious disease tests required by the FDA for plasma donations collected in the United States. Appendix 3 compares EU and U.S. disease testing and donor deferral requirements.
Table 4. FDA Disease Test Requirements for Plasma for
Transfusion and Plasma for Further Manufacturing
Transfusion and Plasma for Further Manufacturing
| Disease | Plasma for Transfusion | Plasma for Further Manufacturinga |
|---|---|---|
| Hepatitis B | Hepatitis B surface Antigen (HBsAg) Hepatitis B core antibody |
HBsAg |
| Hepatitis C | Anti-HCV HCV RNA |
Anti-HCV HCV RNA |
| HIV | Anti-HIV I/II HIV RNA |
Anti-HIV I/II HIV RNA |
| Human T-lymphotropic virus (HTLV) I/II | Anti-HTLV I/II | Not required |
| Syphilis | Serologic test for syphilis, every donation |
Serologic test for syphilis, every 4 months for donors only |
| West Nile virus (WNV)b | WNV RNA | Not required |
|
a
The FDA also encourages in-process NAT testing for parvovirus B19 and hepatitis A. HBV NAT testing also is performed on most Source Plasma.
b
Testing for WNV is recommended in an FDA draft guidance. The FDA is considering recommendations regarding testing for Trypanosoma cruzi (Chagas disease).
|
||
NAT, of which polymerase chain reaction (PCR) is the most widely used form, does not rely on the detection of antibodies produced by the infected host after exposure, but targets the nucleic acid of the infecting agent. By means of the selection of suitable priming molecules (the so-called primers), the assay is highly specific for the infecting virus (see the USP general information chapter Nucleic Acid-Based Techniques—Amplification 1127). Through several cycles of amplification, the polymerase enzyme can repetitively generate copies of the targeted fragment of the viral nucleic acid, providing an exponential amplification of a very short stretch of the viral deoxyribonucleic acid (DNA) [or ribonucleic acid (RNA)]. The exponential amplification leads to the generation of many copies of the target molecule and allows the subsequent detection of this virus-specific fragment, even if the original viral load was exceedingly low. This methodology has brought a new degree of safety.
NAT testing, because of its complexity and expense, is difficult to conduct on individual donations. Generally, aliquots from several donations are combined into a single pool, often called a minipool. Testing in pooled format remains more sensitive than serological ELISA screening of individual donations. In addition, the NAT principle circumvents several of the limitations in detecting pathogens by means of serological methods. Pooling can influence overall sensitivity, depending on the pool size and the analytical sensitivity of the NAT assay employed.
In many countries, the maximal load of a pathogen acceptable for a single donation defines the overall NAT sensitivity required. Assays of higher analytical sensitivity can use larger pools, but those of lower analytical sensitivity must test smaller pools in order to comply with regulations. The availability of commercial NAT test kits with defined analytical sensitivity has made minipools up to 512 very common, because these pool sizes, in combination with the analytical sensitivity of the assays used, comply with common regulations on overall sensitivity.
Effective NAT screening requires that the viral load of the plasma pool at the beginning of production be less than the inactivation and/or removal capacity of the process. Differences between the plasma transfusion and fractionation industries have led to different applications of NAT. For individual donations intended for transfusion, where there is no inactivation and/or removal process and where testing is the only method to interdict a contaminated donation, the safety of each individual donation must be ensured by testing with the most sensitive assays possible. Plasma intended for further manufacturing, in contrast, is pooled and serves as the starting material for a multistep process that has built-in pathogen inactivation methodologies. Therefore, NAT screening for plasma for further manufacture is focused on ensuring safe donations and limiting the viral load of the plasma pool to levels less than the known viral inactivation/removal capacity of the inactivation process.
To avoid the loss of large amounts of plasma from a reactive pool, the fractionation industry has implemented a prescreening strategy, the minipool screening concept mentioned earlier. Aliquots of plasma donations are combined to form minipools, and the minipools are tested by NAT. If a minipool is reactive for a virus tested, the individual donation that gave rise to this positive result can be identified and interdicted. The other donations demonstrated to be free of infection can be used for further manufacturing. The donations are then combined into a production pool, a sample of which is subjected to NAT testing as required by regulations.
As indicated, the NAT test portfolio is not uniform and depends on the intended use of the donation and the regulatory environment. Although screening for HCV RNA is done in most countries, screening for HIV is not universally required. NAT detection of HBV is used mainly for plasma for manufacturing. Screening for B19 virus or hepatitis A virus (HAV) is performed only on plasma for manufacturing. HBV screening using NAT detection is more widespread in European and Asian countries than in the United States.
Donor Deferral
A donor may be deferred from further donation as a result of answers provided on the donor history questionnaire, counseling of donors for reasons for deferral, a medical examination performed at the time of donation, or positive tests for infectious diseases. These processes ensure both the eligibility of the donor and the suitability of the donation. In the event that either is not acceptable, standard processes permanently remove the donor and interdict unused units previously donated. The donor registries mentioned earlier are one means of ensuring that a donor deferred at one center cannot donate elsewhere.
Quarantine and Inventory Hold
Each individual unit of plasma, whether for transfusion or further manufacture, is held in quarantine until all the required tests have been completed. If all required tests have been performed and found acceptable, the unit can be released; if not, the unit must be destroyed. The plasma industry has voluntarily implemented the inventory hold protocol (also discussed in the previous section Donor Screening) for plasma for further manufacture. According to inventory hold requirements, during a 60-day hold period an individual plasma donation cannot be used for further manufacture. The rationale for the hold is that donors who have been recently infected with a pathogen may not have developed levels of antibody at the time of donation, thereby donating an infectious unit despite negative disease tests. The hold provides sufficient time for an infectious donor to develop levels of antibody that will be detected during a subsequent donation if the plasma was intended for transfusion. The 60-day hold also reduces the chance of releasing an infectious unit into the manufacturing process.
The introduction of NAT may have decreased the need for inventory hold, because NAT targets the infecting virus directly and thus does not rely on the time-delayed production of antibodies. Because NAT cannot detect all viruses and because even NAT has a certain (although very low) limit of detection, inventory hold is still of value and thus remains in place in the plasma fractionation industry.
Investigation
Each plasma donation must be traceable from donation to ultimate disposition in order to minimize the potential transmission of an infectious agent. Traceability encompasses all data concerning donation site, donor identifying information, test results, and data regarding transport, storage, and consignee(s).
Look-back is a process to identify and interdict (quarantine) previous donations from a donor who, at a subsequent donation event, has been found to be (1) infected with a transmissible agent or (2) unsuitable for donating plasma because of history, physical examination, or post-donation information. Although look-back strategies are similar in most countries, specific procedures may vary.
QUALITY SYSTEMS
The intent of this section is to outline the general principles and regulations that are the basis of quality systems relating to plasma collection. U.S. collection centers must follow cGMPs that originate in CFR and are elaborated in FDA regulations, guidance documents, and industry standards.
The GMP regulations specifically governing plasma are found in 21 CFR 600, Biological Products: General; and 606, Current Good Manufacturing Practice (cGMP) for Blood and Blood Components. More general cGMP regulations are found in 21 CFR 210, Current Good Manufacturing Practice in Manufacturing, Processing, Packing, or General Holding of Drugs, General; and 21 CFR 211, Current Good Manufacturing Practice for Finished Pharmaceuticals. Although quality systems regulations are part of 21 CFR for medical devices, they have been extended to other manufacturing as part of the “c” in cGMP and FDA Guidance documents.4
The quality system is divided into four major parts: management responsibility, resources, manufacturing operations, and evaluation activities. These are the foundation of the five manufacturing systems: production, facilities and equipment, laboratory control, materials, and packaging and labeling. The procedures of each system are designed to allow operations that facilitate implementation of cGMP requirements. In many instances, these requirements relate to providing facilities and expertise to achieve the requirements for the Five-Layer Safety Net (donor screening, blood testing, donor deferral, quarantine, and investigation), discussed above.
Management responsibilities include providing leadership; building a quality system for the organization that meets requirements; establishing policies, objectives, and plans; and reviewing the quality systems with defined frequency. Resources include having sufficient resources for operational activities, personnel development plans, adequate facilities, and suitable equipment; and controlling outsourced operations. Manufacturing includes designing, developing, and documenting product and processes, performing and monitoring operations, and addressing nonconformities. Evaluation activities include analyzing data for trends, conducting internal audits, and initiating corrective and preventive actions.
A number of required routine activities related to collection and release of plasma or normal recovered plasma from whole blood are linked to both cGMP guidelines and quality systems. These include the requirement for having SOPs to cover all aspects of collection, testing, and release. It is also necessary to validate equipment and systems used by the collection center, including temperature-controlled areas, laboratory equipment, water systems, and computer systems. The design and operation of the facility must be adequate to perform the tasks at hand and prevent cross-contamination. Plasma collection facilities must have an adequate number of knowledgeable and trained staff as well as procedures for acceptance and release of raw materials. To the extent possible, collection facilities must adhere to the GMP requirements for a pharmaceutical manufacturing facility.
GLOSSARY
Apheresis—A method of obtaining one or more blood components by machine processing of whole blood; the residual components of the blood are returned to the donor during or at the end of the process.
Blood component—A constituent of human blood: red cells, white cells, platelets, or plasma.
Blood establishment—Any structure or organization responsible for any aspect of the collection and testing of human blood or blood components, whatever their intended purpose, and their processing, storage, and distribution. Hospital transfusion services engaged only in compatibility testing and transfusion of blood and blood products are not included within the definition of blood establishment.
Blood product—Any therapeutic product derived from human blood or plasma.
Center—Collection site or location where blood or plasma is collected (and also may be processed and stored). Center is also applicable to a testing Site (see entry in this glossary).
Cryoprecipitate—A plasma component prepared from fresh-frozen plasma by freeze-thaw precipitation of proteins and subsequent concentration and resuspension of the precipitated proteins in a small volume of the plasma.
Deferral—Temporary or permanent suspension of the eligibility of an individual to donate blood or blood components.
Distribution—The act of delivery of blood and blood components to other blood establishments, hospital blood banks, and manufacturers of blood products.
Manufacturing pool—A combination of a specified number of plasma donations used as the first step in the manufacture of plasma derivatives.
Donation minipool—A combination of a small number of units or samples representative of donations used for pretesting prior to pooling units for manufacture.
Quarantine—The physical isolation of blood components or incoming materials/reagents over a variable period of time while awaiting acceptance, issuance, or rejection of the blood components or incoming material/reagents.
Site—Any location at which a blood establishment carries out blood collection, not including any location not owned or managed by the blood establishment at which blood is collected or any mobile blood collection unit.
Validation—The establishment of documented and objective evidence that the particular requirements for a specific intended use can be consistently fulfilled.
ABBREVIATIONS
| AABB | American Association of Blood Banks |
| AIDS | Acquired immune deficiency syndrome |
| CFR | Code of Federal Regulations |
| CJD | Creutzfeldt-Jakob disease |
| CMV | Cytomegalovirus |
| CNS | Central nervous system |
| DNA | Deoxyribonucleic acid |
| EBV | Epstein-Barr virus |
| EIA | Enzyme immunoassay |
| EU | European Union |
| FBS | Fetal bovine serum |
| FDA | Food and Drug Administration |
| FFP | Fresh-frozen plasma |
| FP24 | Plasma frozen within 24 hours after phlebotomy |
| cGMP | Current Good Manufacturing Practice |
| HAV | Hepatitis A virus |
| HBsAg | Hepatitis B virus surface antigen |
| HBV | Hepatitis B virus |
| HCT | Hematocrit |
| HCV | Hepatitis C virus |
| HIV | Human immunodeficiency virus |
| HLA | Human leukocyte antigen |
| HTLV | Human T-lymphotropic virus |
| IgA, IgG, IgM | Immunoglobulins A, G, and M, respectively |
| IQPP | International Quality Plasma Program |
| IU | International Unit |
| NAT | Nucleic acid amplification technology |
| NDDR | National Donor Deferral Registry |
| PCR | Polymerase chain reaction |
| PPTA | Plasma Protein Therapeutics Association |
| PRP | Platelet-rich plasma |
| RNA | Ribonucleic acid |
| SARS | Severe acute respiratory syndrome |
| TEP | Therapeutic exchange plasma |
| WBDP | Whole blood-derived plasma |
| WNV | West Nile virus |
APPENDICES
Appendix 1
Note—The collection, processing, and uses of plasma have generated a large number of terms and definitions that reflect the diversity of operations. In addition to the FDA standards and terms, industrywide voluntary standards are discussed in the Plasma Safety Considerations section.
Advisory Note: These terms are not meant as regulatory definitions, because plasma term definitions can vary from region to region and among industry sectors. The reader is advised to consult with regulatory authorities responsible for the region and industry sector. Often specific process variables must be considered.
Appendix 1: Plasma Types and Specifications as Assigned by Regulatory Agencies in Selected Jurisdictions
| Plasma Type and Agency or Agency Type | Specification | |
|---|---|---|
| Recovered plasma | ||
| Plasma derived from single units of whole blood as a by-product in the preparation of blood components from whole blood collection and intended for further manufacturing. Compliance Policy Guides Manual (CPG 7134.12), Sec. 230.100. | ||
| Plasma for use in manufacturing and prepared from allogenic donations. Plasma selected for manufacture that has been collected from whole blood or apheresis plasma collected for transfusion that has expired. | ||
| Plasma separated from whole blood most often by manual centrifugation or by apheresis. The priority for the blood collected is usually for the production of red blood cells. However, the plasma can be suitable for further manufacture of biotherapeutics and transfusion. The time from collection to freezing can vary depending on the distance of collection and processing sites. Volunteer donors typically are used. | ||
| Source Plasma | ||
| Fluid portion of human blood collected by plasmapheresis and intended as source material for further manufacturing use (21 CFR 640.60). | ||
| Plasma separated from whole blood by plasmapheresis where the cellular components can be returned to the donor. The priority for the plasma usually is for further manufacture of biotherapeutic products. However, the plasma can be suitable for transfusion. It is rapidly frozen after collection. | ||
| Fresh-frozen plasma (FFP) | ||
| Fresh-frozen plasma shall be prepared from blood collected by a single uninterrupted venipuncture with minimal damage to and minimal manipulation of the donor’s tissue. The plasma shall be separated from the red blood cells and placed in a freezer within 8 hours or within the timeframe specified in the directions for use for the blood collecting, processing, and storage system and stored at |
||
| Plasma separated from the blood of an individual donor and placed at |
||
| Plasma that is collected and frozen quickly after preparation. Transfusion is the primary intended use. However, FFP can be suitable for further manufacture of biotherapeutic products. | ||
| A component for transfusion or for fractionation prepared either from whole blood or from plasma collected by apheresis, frozen within a period of time and to a temperature that will adequately maintain labile coagulation factors in a functional state (Chapter 21). | ||
| Supernatant plasma separated from a whole blood donation or plasma collected by apheresis, frozen, and stored. | ||
| Plasma, Fresh Frozen is a component for transfusion or for fractionation prepared either from whole blood or from plasma collected by apheresis, frozen within a period of time and to a temperature that will adequately maintain the labile coagulation factors in a functional state. If prepared from whole blood, it should preferably be recovered within 6 hours, and not more than 18 hours after collection if the unit has been refrigerated. Plasma may also be collected up to 24 hours if the collected blood has been immediately cooled and maintained at 20 |
||
| Concurrent plasma | ||
| Plasma collected concurrently with cellular components. Concurrent plasma may be suitable for transfusion or for further manufacture of biotherapeutics. | ||
| Applicant Donor | ||
| Source Plasma obtained during the first collection from a new donor. The plasma is reserved for testing, and any remainder or products derived from the remainder are not allowed for use in humans or are quarantined until the donor passes appropriate tests and returns for a second donation which also clears testing. At that time, both collections are reclassed as “Qualified”. | ||
| Platelet-rich plasma (PRP) | ||
| PRP shall be prepared from blood collected by a single uninterrupted venipuncture with minimal damage to and manipulation of the donor’s tissue. The plasma shall be separated from the red blood cells by centrifugation within 4 hours after completion of the phlebotomy or within the timeframe specified in the directions for use for the blood collecting, processing, and storage system. The time and speed of the centrifugation shall have been shown to produce a product with at least 250,000 platelets per µL. The plasma shall be stored at a temperature between 20 |
||
| Plasma that is a product of the first centrifugation of blood where it is separated from red cells. Platelets are fractionated into the plasma layer. | ||
| Platelet-poor plasma | ||
| Plasma that is further purified from platelets by a second centrifugation of PRP. | ||
| Cryo-poor plasma | ||
| Plasma that remains after both platelets and cryoprecipitated AHF have been removed may be labeled “Plasma, Cryoprecipitate Reduced” [21CFR 640.34(e)(2)]. | ||
| Plasma Cryoprecipitate Reduced; Fresh-frozen Plasma from which cryoprecipitate has been removed. | ||
| Plasma that has been thawed by maintaining the temperature just above freezing (usually 4 |
||
| Plasma, Fresh-Frozen, Cryoprecipitate-Depleted (Chapter 23). A component prepared from plasma by the removal of cryoprecipitate. | ||
| Plasma cryoprecipitate-depleted for transfusion means a plasma component prepared from a unit of plasma, fresh-frozen. It comprises the residual portion after the cryoprecipitate has been removed. | ||
| Plasma, Fresh Frozen, Cryoprecipitate-Depleted is a component prepared from Fresh Frozen Plasma by the removal of cryoprecipitate. The content of albumin, immunoglobulin, and most clotting factors is maintained, but the levels of Factors V and VIII and fibrinogen are reduced. It can be stored for up to 36 months at below |
||
| Cryo-rich plasma | ||
| Plasma that has been thawed by gentle heat input (e.g., in a 37 |
||
| Plasma for labile products | ||
| Plasma that has been collected and best maintains the activity and integrity of labile plasma proteins as exemplified by clotting Factor VIII. Generally the time from collection through processing to freezing is rapid. | ||
| Plasma for stable products | ||
| Plasma that has been collected where conditions for preservation of labile products was not achieved, however conditions were sufficiently moderate so relatively stable products like IgG and albumin would not be impacted. | ||
| Less than 6-hour plasma | ||
| This is generally recovered plasma that has been collected, processed, and frozen prior to 6 hours after collection. This plasma generally is considered to be acceptable for the production of labile products. | ||
| 6- to 12-hour plasma | ||
| Generally this is recovered plasma that has been collected, processed, and frozen more than 6 hours and less than 12 hours after collection. This plasma is generally considered to be acceptable for the production of labile products but is inferior to less than 6-hour plasma for this purpose. | ||
| 12- to 24-hour plasma | ||
| Generally this is recovered plasma that has been collected, processed, and frozen more than 12 hours and less than 24 hours after collection. This plasma may be acceptable for the production of labile products but is inferior to less than 6-hour plasma and 6- to 12-hour plasma for this purpose. | ||
| Less than 12-hour plasma | ||
| Generally this is recovered plasma that has been collected, processed, and frozen less than 12 hours after collection. This plasma may be acceptable for the production of labile products but is inferior to less than 6-hour plasma and 6- to 12-hour plasma for this purpose. | ||
| More than 24-hour plasma | ||
| Generally this is recovered plasma that has been collected, processed, and frozen more than 24 hours after collection and usually less than 72 hours after collection. This plasma generally is not acceptable for the production of labile products. | ||
| Pooled plasma | ||
| Plasma that has been pooled for manufacturing from several donors. Some plasma pools for further manufacture of biotherapeutic products can be derived from several hundred to a few thousand donors. | ||
| Single-donor plasma | ||
| Plasma derived from a single donor. It can be a single unit or a pool of several units derived from multiple collections from the same donor. | ||
| Hyperimmune plasma | ||
| Plasma derived from donors with high titers to specific disease agents. Titers are elevated in these donors mostly as a result of immunization with a vaccine (e.g., hepatitis B, tetanus, or rabies) or exposure to disease agents (e.g., HCV or SARS). Hyperimmune plasma usually is intended for the preparation of IgG to provide passive immunity against target disease agents. | ||
| S/D plasma | ||
| Plasma that has been treated with solvent/detergent, an inactivation method effective against envelope virus disease agents (e.g., HIV, HBV, or HCV). Some plasma protein components are inactivated or damaged by the process (e.g., alpha-1 proteinase inhibitor, Protein S, anti-plasmin, or FVIII). | ||
| Human Plasma Pooled and Treated for Virus Inactivation is a frozen or freeze-dried, sterile, nonpyrogenic preparation obtained from human plasma derived from donors belonging to the same ABO blood group. The preparation is thawed or reconstituted before use to give a solution for infusion. The human plasma used complies with the monograph on Human Plasma for Fractionation. | ||
| Therapeutic exchange plasma (TEP) | ||
| Similar to Source Plasma in its collection. However, the donors are patients who are having their plasma replaced with electrolytes, protein solutions, or plasma from another donor. The objective usually is to remove disease elements from the patient’s plasma. Generally, TEP is not advisable for further manufacture of biotherapeutic products. However, there may be cases where a specialty product may propose a specific TEP as a source material. | ||
| Quarantine plasma | ||
| Plasma that has been collected and not had initial testing completed and/or stored as part of a controlled donor program. The donor is retested for disease agents (e.g., 6 months after collection). If the donor again is negative for the tested disease agents, then the plasma is released for use for further manufacture and/or use in humans. | ||
| Quarantine residual plasma | ||
| Plasma that has been collected and stored as part of a controlled donor program. The donor was not retested for disease agents (e.g., 6 months after collection). An example would be that the donor did not return to the collection facility to permit the later test. Quarantine Residual Plasma is not recommended for further manufacture and/or use in humans. | ||
| Salvaged plasma | ||
| Plasma that has experienced a storage or transport temperature deviation but may still be useful for the preparation of nonlabile products such as albumin or IgG. | ||
| Plasma for fractionation (Processing requirements) | ||
| Placed in a freezer within 8 hours and stored at |
||
| The liquid part of human blood remaining after separation of the cellular elements from blood collected in a receptacle containing an anticoagulant or separated by continuous filtration or centrifugation of anticoagulated blood in an apheresis procedure. It is intended for the manufacture of plasma-derived products. When the plasma is intended for the recovery of proteins that are labile in plasma, it is frozen rapidly to |
||
| Human plasma for fractionation is the liquid part of human blood remaining after separation of the cellular elements from blood collected in a receptacle containing an anticoagulant or separated by continuous filtration or centrifugation of anticoagulated blood in an apheresis procedure. It is intended for the manufacture of plasma-derived products. When obtained by plasmapheresis or from whole blood (after separation from cellular elements), plasma intended for the recovery of proteins that are labile in plasma is frozen within 24 hours of collection by cooling rapidly in conditions validated to ensure that a temperature of When obtained by plasmapheresis, plasma intended solely for the recovery of proteins that are not labile in plasma is frozen by cooling rapidly in a chamber at When obtained from whole blood, plasma intended solely for the recovery of proteins that are not labile in plasma is separated from cellular elements and is frozen in a chamber at Frozen plasma is stored and transported in conditions designed to maintain the temperature at or below |
||
| Plasma frozen within 24 hours after phlebotomy | ||
| Plasma manufactured from whole blood should be frozen within 24 hours after phlebotomy. Blood component must be labeled “Plasma Frozen Within 24 Hours after Phlebotomy.” |
Appendix 2: Donor Criteria
| Region | ||||||||
|---|---|---|---|---|---|---|---|---|
| Criterion | United States | United Kingdom | European Union | Australia | ||||
| General criteria for blood donation | ||||||||
| Appearance | Donor should appear to be in good health (AABB) |
Donor should be in good health | Only donors in good health accepted | Nothing specific noted | ||||
| Underlying medical conditions | Most serious medical conditions are grounds for deferral under United States, EU, and UK guidelines. FDA guidelines require deferral only if a person has used bovine insulin manufactured from UK cattle. However, AABB guidelines require deferral for cancer, heart, liver, or lung disease, and bleeding tendency unless approved by a medical director. Cancer and cardiac disease and diabetes treated with insulin require permanent deferral under EU and UK guidelines | Most serious medical conditions are grounds for deferral. Cancer: accepted 5 years of remission. Cardiac disease: varies depending on clinical condition. Diabetes: acceptable if controlled. |
||||||
| Age | state law (AABB, whole blood) |
Between 18 and 65; donation at 17 permitted if in accord with national legislation; first-time donors >60 only if permitted by physician | Whole blood: between 17 and 65; no first-time donors >60 Apheresis: first-time donors between 18 and 60; may donate up to age 65 |
Whole blood: can start at 16–17 with consent of parents and continue to 80, but medical review required at >70. Apheresis: accept new donors 18–65, with medical evaluation required when >60. Existing donors require annual medical review if >65. |
||||
| Weight | None stated: no more than 10.5 mL/kg may be withdrawn (AABB) FDA requires Source Plasma donors to weigh at least 110 lb |
Whole blood: Apheresis: no specific weight requirement |
Whole blood: Apheresis: |
<45 kg—defer. Medical opinion required for unexplained weight loss |
||||
| Blood pressure | Systolic Diastolic (AABB) |
Systolic Diastolic |
No specified blood pressure parameters; donors with high blood pressure may donate provided (1) they have not suffered any complications caused by high blood pressure, (2) they are taking only beta blockers and/or diuretics, and (3) their disease is stable, as determined by qualified medical personnel. | Acceptable ranges: Systolic 90–180 mm Hg. Diastolic 60–90 mm Hg. Hypertension 180–100: defer. Hypotension 90–60: defer. |
||||
| Pulse | Between 50 and 100 beats per minute and regular; lower pulses acceptable at discretion of physician (AABB) | Between 50 and 100 beats per minute and regular | No specific pulse rate parameters stated | Regular pulse between 50 and 100: accept Pulse between 40 and 49: accept if donor is physically fit and not on medication |
||||
| Temperature | Donors who have had a temperature of |
Donors who have had a temperature of |
No requirement | |||||
| HgB/HCT | Males: Females: |
Males: Females: |
Whole blood: females, 120–165 g/L; males, 130–185 g/L. Apheresis: females, 115–165 g/L; males, 125–185 g/L. If high, defer. If low, defer for 6 months and test for ferritin. |
|||||
| Skin examination | Free of infectious skin disease at site of phlebotomy; no skin punctures or scars indicative of addiction to self-injected narcotics (CFR) Free of infectious diseases (AABB) |
Skin at venipuncture site should be clear of lesions, including eczema | There should be no skin disease at venipuncture site | Avoid venesection where there is evidence of inflammation or infection | ||||
| Pregnancy | Defer for 6 weeks after delivery (AABB) | Defer 6 months after delivery | Defer for 1 week for every completed week of pregnancy | Current: defer 9 months from estimated date of confinement. After third-trimester delivery: defer 9 months. Miscarriage or termination: defer 3, 6, or 9 months, respectively for 1st, 2nd, and 3rd trimester. |
||||
| Underlying medical conditions that require deferral and that do not pose a risk of transfusion-transmissible infection | ||||||||
| Cancer | Permanent deferral unless deemed suitable by medical director (AABB) | Permanent deferral although physician may make exceptions. Permitted after cervical cancer or basal cell carcinoma if successfully treated | Malignant neoplasms, including leukemias and myeloproliferative disorders, are cause for permanent deferral; exceptions may be made for certain conditions after successful therapy | Permanent deferral for haematological malignancies. Skin cancer–basal–cell carcinoma: accept. Other cancers: defer 5 years after completion of treatment |
||||
| Cardiac disease | Free of major organ disease (heart, liver, and lungs) unless deemed suitable by medical director (AABB) | Permanent deferral for persons with a history of heart disease, especially coronary disease, angina pectoris, severe cardiac arrhythmia, arterial thrombosis, or recurrent venous thrombosis. 2-year deferral for rheumatic heart disease with no evidence of chronic disease | Permanent deferral for persons with active or past serious cardiovascular disease, except congenital abnormalities with complete cure | Permanent deferral for arrhythmias, endocarditis, ischaemic heart disease, heart surgery, myocardial disease. Accept: congenital heart disease if surgically corrected. Heart murmurs: accept, subject to medical opinion. Accept after full recovery: pericardial disease, rheumatic heart disease. |
||||
| Cerebrovascular diseases | No specific guideline other than that the donor must be free of major organ disease (AABB) | Permanent deferral for history of cerebrovascular diseases | Permanent deferral for donors with a history of serious CNS disease | Permanent deferral | ||||
| Epilepsy | No deferral | Must be free of epileptic attack for 3 years and have been taken off all medication | Permanent deferral unless at least 3 years have elapsed since the date that donor last took anticonvulsant medication and there has been no recurrence of symptoms | Defer for 2 years from last seizure | ||||
| Gastrointestinal disease | No specific deferral; free of major organ disease unless deemed suitable by medical director (AABB) | No specific deferral | Deferral (not noted to be a permanent deferral) for disease that renders the individual liable to impaired iron absorption or blood loss | Ulcers: defer indefinitely | ||||
| Genitourinary and renal disease | No specific deferral. Free of major organ disease unless deemed suitable by medical director (AABB) |
Five-year deferral after complete recovery from acute glomerulonephritis | Permanent deferral for donor with serious genitourinary or renal disease | Permanent deferral: chronic pyelonephritis, chronic kidney infection, chronic dialysis. Accept if resolved: haematuria, acute kidney infection. Urinary catheter present: plasma only for fractionation if underlying condition acceptable. Acute dialysis: defer for 12 months. Acute glomerulonephritis: defer 5 years after recovery. |
||||
| Diabetes | No specific deferral except that receipt of bovine insulin manufactured in the UK requires permanent deferral (FDA). Free of major organ disease unless deemed suitable by medical director |
Permanent deferral if insulin therapy required | Permanent deferral for donors on insulin treatment | Permanent deferral if diabetes-associated complications present Accept if disease is controlled, even if patient is taking insulin |
||||
| Respiratory disease | Free of acute respiratory disease (CFR). Free of major organ disease (lungs) unless deemed suitable by medical director (AABB) |
Permanent deferral for chronic bronchitis; common cold acceptable | Permanent deferral for serious disease | Permanent deferral: chronic abscess, bronchiectasis, or emphysema with respiratory insufficiency. Accept if mild and controlled: asthma, bronchiectasis without respiratory insufficiency Plasma for fractionation only: chronic bronchitis. Acute bronchitis: defer 2 weeks after recovery and being off antibiotics for 5 days. Pleurisy, pneumonia: defer 4 weeks after recovery. Acute pulmonary embolism: defer 12 months after recovery |
||||
| Hematologic disorders | Free of abnormal bleeding tendency unless determined suitable by medical director (AABB) Free of major organ disease (cancer) unless deemed suitable by medical director (AABB) |
No specific deferral. Donors who are heterozygous for beta thalassemia eligible if HgB values are within normal limits | Permanent deferral for donors with serious hematologic diseases | Permanent deferral: donors with serious hematologic diseases (sickle cell disease, thalassaemia major). Accept: thalassaemia minor. Defer: anaemia Plasma for fractionation only: elliptocytosis, glucose-6-phosphate dehydrogenase (G6PD) deficiency, spherocytosis. Apheresis only permitted for patients with G6PD deficiency |
||||
| Immunologic disorders | No specific deferral. Free of major organ disease unless deemed suitable by medical director (AABB) |
Permanent deferral for donors with autoimmune disease that affects more than one organ. Defer: documented history of anaphylaxis |
Permanent deferral for donors with serious immunologic diseases | Permanent deferral: autoimmune disorders. Accept: If asymptomatic, only one organ system involved, not on immunosuppressive therapy. Accept: Sjogren’s syndrome |
||||
| Metabolic disease | No specific deferral. Free of major organ disease unless deemed suitable by medical director (AABB) |
No specific deferral | Permanent deferral for serious metabolic disease | No specific reference | ||||
| Bone disease | No specific deferral. Free of major organ disease unless deemed suitable by medical director (AABB) |
Two-year deferral after having been declared cured of osteomyelitis | No specific deferral | No specific reference | ||||
| Surgery | No specific deferral unless blood was transfused; in which case, a 12-month deferral applies (CFR) | Major surgery requires evaluation of risk for transfusion-transmissible disease | Permanent deferral for persons with history of resection of the stomach. Major surgery requires a 6-month deferral; minor surgery requires a 1-week deferral | Minor (e.g., skin lesions, arthroscopy): defer until recovered. Routine minor (e.g., appendectomy, laparoscopy): defer for 2 months. Major surgery (if donor received autologous blood only): defer 6 months. Neurosurgery; medical assessment required. |
||||
| Medications that require deferral | ||||||||
| Antibiotics | As defined by medical director (AABB) | Donors treated with any prescribed drug should be deferred for a period consistent with the pharmacokinetic properties of the drug | Defer for 2 weeks from full recovery or 1 week from cessation of antibiotic therapy, whichever is longer | Acute treatment: defer until recovered and off antibiotics for 5 days. Prophylactic: accept plasma only for fractionation. Topical: accept if skin is unbroken and not infected. |
||||
| Drugs with teratogenic potential |
Etretinate: permanent deferral (FDA). Acitretin: 3-year deferral from last dose (FDA). Dutasteride: 6-month deferral from last dose (FDA). Isotretinoin: 1-month deferral from last dose (FDA). Finasteride: 1-month deferral from last dose (FDA). |
Donors treated with drugs with proven teratogenic effect should be deferred for a period consistent with the pharmacokinetic properties of the drug | Donors taking drugs that are proven or potential teratogens or who are taking drugs that accumulate in the tissues over long periods of time should not be used as blood donors | Raloxifene (Evista): defer for 6 months after completion of treatment. Acitretin (Neotigason): defer for 3 years after completion of treatment. Etretinate (Tigason): permanent deferral. Finasteride (Proscar): 7 days after completion of treatment. Isotretinoin: defer for 8 weeks after completion of treatment. |
||||
| Growth hormone from human pituitary glands | Permanent deferral (FDA) | Permanent deferral | ||||||
| Other drugs | No other deferrals by FDA or AABB. Any other deferral at discretion of medical director of blood center (AABB). Warfarin: 7-day deferral for plasma donation (AABB). |
Recommended that a list of commonly used drugs with rules for acceptability of donors, approved by the medical staff of the transfusion center, be available. | Other drugs acceptable as long as the underlying condition for which the drug is taken is acceptable. | Most medications that are taken by donors are not harmful to recipients; therefore people taking medications can be acceptable as blood donors. Eligibility is based on the assessment of the underlying condition and specific medication guidelines. | ||||
| Insulin | Permanent deferral: bovine insulin made in UK (FDA). | Permanent deferral if treated with insulin | Permanent deferral if treated with insulin | Defer if diabetes is poorly controlled | ||||
| Immunizations | ||||||||
| Toxoids | No deferral | No deferral | No deferral | |||||
| Licensed killed bacterial vaccines | No deferral | No deferral | No deferral | |||||
| Licensed inactivated viral vaccines | No deferral | No deferral | No deferral except 1-week deferral after hepatitis B vaccination | No deferral except 1-week deferral after hepatitis B vaccination | ||||
| Unlicensed killed vaccines | 1-year deferral (AABB) | Defer 3 months after vaccination | ||||||
| Inactivated rickettsial vaccines | No deferral | No deferral | ||||||
| Live attenuated bacterial and viral vaccines | 4 weeks for varicella and rubella 2 weeks for rubeola, yellow fever, mumps, polio (oral), typhoid (oral) (AABB) |
4 weeks | 8 weeks | Plasma only for fractionation for 4 weeks after vaccination | ||||
| Transfusion-transmissible infections that require deferral | ||||||||
| HIV infection/AIDS and sexual partners | Permanent deferral if present or past clinical or laboratory evidence of HIV infection/AIDS: positive EIA with positive or indeterminate confirmatory test; positive NAT test; clinical signs include unexplained weight loss, night sweats, blue or purple spots in mouth or on skin, white spots or unusual sores in the mouth, swollen lymph nodes for more than 1 month, persistent cough or shortness of breath, persistent diarrhea, fever for more than 10 days; sexual partners deferred for 1 year from time of last contact (FDA) | Permanent deferral for donors found to have a confirmed positive marker for HIV. Donors found to have a repeat positive marker for HIV that cannot be confirmed should be informed according to the nationally agreed algorithm. |
Donors with HIV I or II must be permanently deferred | Infection: permanent deferral. Relevant symptoms within the last 6 months: defer for 12 months. Sexual contact with HIV-positive partner: defer for 12 months after last sexual contact. |
||||
| Hepatitis and sexual partners and household contacts | Permanent deferral for the following: History of viral hepatitis after the 11th birthday. Confirmed repeatedly reactive for HBsAg. Positive test for anti-HBc on more than 1 occasion (testing is not required for Source Plasma donors). Present or past laboratory or clinical evidence of infection with HCV. Sexual partners of patients with hepatitis deferred for 1 year from last contact; household contacts of persons with hepatitis B deferred for 1 year from last contact (CFR). |
Permanent deferral for donors whose blood gives a positive reaction for the presence of HBsAg and/or anti-HCV. Donors with a history of jaundice or hepatitis may, at the discretion of the appropriate competent medical authority, be accepted as blood donors, provided that an approved test for HBsAg and anti-HCV is negative. |
Permanent deferral for hepatitis B and C: donors with history of hepatitis B may donate after 12 months after recovery, provided that all markers are negative or core antibody positive, HBsAg is negative, and anti-HbS |
Hepatitis B acute or past infection: defer for 12 months after recovery, then perform hepatitis testing. Hepatitis B chronic carrier: permanent deferral. Hepatitis B contact, sexual, mucosal, household: defer for 12 months from last exposure unless immune. Hepatitis B other contact: accept. Hepatitis C positive past infection: permanent deferral. Hepatitis C contact, sexual, mucosal, household: defer for 12 months from last exposure; other contact: accept. |
||||
| HTLV | Present or past clinical or laboratory evidence of infection with HTLV I/II (positive EIA on 2 occasions). [HTLV not tested in Source Plasma donors] (FDA) |
Permanent deferral for carriers of HTLV I/II | Permanent deferral for donors with HTLV I/II | Infection: permanent deferral. Repeat reactive status: plasma only for fractionation. Sexual contact: defer for 12 months after last contact. Household contact: accept. |
||||
| West Nile virus | Donors with symptoms suspicious of or actual diagnosis of WNV deferred for 120 days. Donor testing positive on WNV NAT deferred for 120 days. Donor who develops symptoms of WNV within 2 weeks of donation should be deferred for 120 days. Donors implicated in possible transfusion-transmitted WNV infection should be deferred for 120 days (FDA). | Defer for 28 days after donor leaves an area with ongoing transmission to humans. | Defer for 6 months if donor was in area endemic for WNV and was diagnosed with or had symptoms consistent with WNV. Defer for 28 days after donor returns from endemic area, provided donor has no symptoms of WNV. |
Infection: defer for 3 months after full recovery. Area exposure: plasma only for fractionation for 8 weeks after leaving risk area. |
||||
| Chagas disease and babesiosis |
Permanent deferral for history of Chagas disease; current FDA regulations do not require testing for Chagas disease. However, although not required, most facilities collecting blood for transfusion perform EIA test for Chagas and permanently defer following positive test. (AABB) FDA probably will not require antibody screening for fractionated or recovered plasma used for further manufacture. FDA has granted exemptions and permitted the collection and distribution of Source Plasma for further manufacture into noninjectable products from a donor known to have Chagas disease (CFR). Permanent deferral for history of babesiosis (AABB) |
Permanent deferral for individuals with Chagas disease or history of Chagas disease; blood of persons who were born or have been transfused in areas where the disease is endemic should be used only for plasma fractionation products unless a validated test for infection is negative | Permanent deferral for individuals with Chagas disease. Individuals in the following categories may donate 6 months after leaving an endemic area, provided that a validated test for Chagas disease is negative (if a validated test is positive or not performed, the donor is permanently deferred): born in South or Central America, mother born in South or Central America, transfused in South or Central America; lived or worked in a rural subsistence community in South or Central America for 4 weeks or more | Infection: permanent deferral. Contact: accept. Chagas disease area resident: plasma only for fractionation, permanently. Chagas disease area visitor: plasma only for fractionation for 12 months after leaving endemic area |
||||
| Creutzfeld-Jakob disease (CJD) and variant CJD | Permanent deferral if donor: has diagnosis of CJD or nvCJD; has a blood relative diagnosed with CJD; received dura mater graft; received human-derived pituitary growth hormone; received bovine insulin made in UK; spent a cumulative 3 months in UK between 1980 and 1996; received a blood transfusion in the UK at any time since 1980; spent 6 months between 1980 and 1990 on a US military base in Northern Europe; spent 6 months between 1980 and 1996 on a US military base elsewhere in Europe; spent a cumulative 5 years in Europe (Source Plasma donors are not deferred for the latter) (FDA) |
Permanent deferral if donor: treated with extracts derived from human pituitary glands; has been recipient of dura mater or corneal graft; has a family risk of CJD or any other TSE. For vCJD: Member states should determine on the basis of the prevalence of BSE within individual countries, of the endogenous exposure of the population to bovine products imported from countries with a high BSE prevalence, and of the incidence of cases of vCJD, what precautionary measures they may need to take to minimize the risk of transmission of vCJD via blood transfusion |
Permanent deferral if donor: Diagnosed with CJD, vCJD, or any other prion-associated disease; has family risk of CJD; at increased risk from surgery, transfusion, or transplant of tissues or organs; received a dura mater graft; received a corneal, scleral, or ocular graft; received human-derived pituitary extract; received a blood transfusion in UK since 1980; received intravenous immunoglobulin (IVIg) of UK origin; donated unit of blood implicated in possible case of transfusion-related vCJD. Additionally, all plasma from British donors cannot be used for fractionation |
Permanent deferral if diagnosed with any prion-related disease. Permanent deferral if donors have spent a cumulative time of 6 months in England, Wales, Scotland, Northern Ireland, the Channel Islands, or the Isle of Man between 1 January 1980 and 31 December 1996. Permanent deferral if donors received a blood transfusion or blood products in England, Wales, Scotland, Northern Ireland, the Channel Islands, or the Isle of Man from 1 January 1980 onwards unless the blood products were processed plasma products and were given after 31 December 2001. Permanent deferral if the donor had ear surgery performed between 1972 and 1989 and dura mater was used. Permanent deferral for donors who received human-derived pituitary growth and gonadotrophic fertility hormones prior to 1986. |
||||
| Visceral leishmaniasis (Whole Blood) |
Donors who have been to Iraq are deferred for 1 year. Permanent deferral for signs and symptoms of visceral leishmaniasis (FDA) | No specific deferral | Permanent deferral for visceral leishmaniasis | Cutaneous: plasma only for fractionation, permanently Visceral: permanent deferral Contact: accept. |
||||
| Medical conditions and behaviors that place an individual at risk for a transfusion-transmissible infection and require deferral. | ||||||||
| Xenotransplant | No current deferral required for blood donation, but draft regulations for tissue and organs require permanent deferral. | Permanent deferral | Permanent deferral | Permanent deferral | ||||
| Blood transfusion organ and tissue transplant; treatment with plasma-derived clotting factor concentrates | One-year deferral (dura mater graft is permanent deferral); permanent deferral if patient received clotting factor concentrates and sexual partner deferred for 1 year after last contact | Six-month deferral: if NAT test for hepatitis C is negative, may donate after 4 months | Permanent deferral if donor ever received clotting factor concentrate or was transfused after 1 January 1980; 1 year for tissue or organ transplant | Blood transfusion homologous: defer 12 months Coagulation factor, blood derived, short term: defer 12 months from last treatment. Coagulation factor, blood derived, continuous: permanent deferral. Human tissue recipients: Organ/haematological: permanent deferral. Homologous, bone, tendon, skin: accept. Collagen: accept. Corneal: permanent deferral for iatrogenic cCJD risk. |
||||
| Surgery or use of endoscope with biopsy | No specific deferral criteria; general health and transfusion criteria apply | Six-month deferral for major surgery; stomach resection requires permanent deferral; 1-week deferral for minor surgery; 6-month deferral for endoscope with biopsy; if NAT test for hepatitis C is negative, may donate after 4 months | Six-month deferral for major surgery or procedure using an endoscope; 1-week deferral for minor surgery | Defer 6 months | ||||
| Nonsterile skin penetration or mucous membrane exposure to blood or body fluids not the donor’s own | Twelve-month deferral | Six-month deferral; if NAT test for hepatitis C is negative, may donate after 4 months | Twelve-month deferral | Defer 12 months | ||||
| Acupuncture, tattoo, body piercing, etc. | Twelve-month deferral unless performed by a state-regulated entity, using sterile needles and disposable dyes | Six-month deferral; if NAT test for hepatitis C is negative, may donate after 4 months; exception can be made according to national risk assessment | Twelve-month deferral; 6-month deferral if validated test for hepatitis B core antibody is negative; for acupuncture, no deferral if performed by state-regulated entity | If using single-use items: plasma only for fractionation for 12 months If not single-use or unsure: defer for 12 months |
||||
| Injection of medications or steroids not prescribed by a physician | Permanent deferral; sexual partner deferred for 1 year | All blood donors should be provided with accurate and updated information about HIV transmission and AIDS so that persons who have unsafe sex practices or other risk behavior exposing them to potential infection will refrain from donating. The information provided may vary among countries according to local epidemiological data. | Permanent deferral if donor ever injected or has been injected with drugs; sexual partner deferred for 1 year | Permanent deferral | ||||
| Males who have sexual contact with another male | Permanent deferral for sexual contact, even once, since 1977; female sexual partner deferred for 1 year from last contact | Permanent deferral for oral or anal sexual contact even if protection used; female sexual partner deferred for 1 year from last contact. | Defer for 12 months after last sexual contact | |||||
| Accepted money or drugs or other payment in exchange for sex since 1977 | Permanent deferral; sexual partner deferred for 1 year from last contact | Permanent deferral; sexual partner deferred for 1 year from last contact. | Defer for 12 months after last sexual contact | |||||
| Incarceration for more than 72 hours in the past year | One-year deferral | No deferral | Defer for 12 months after release | |||||
| Born in or lived in Africa | Permanent deferral if born or lived in countries where HIV I subtype O is endemic (Cameroon, Central African Republic, Chad, Congo, Equatorial Guinea, Gabon, Niger, Nigeria); sexual partner deferred for 1 year from last contact unless tested with a test validated to detect Group O | No specific deferral for Africa; malaria rules apply; however, sexual partners of persons who were sexually active in areas where HIV is endemic deferred for 1 year from last contact | No specific deferral: donors who have visited a malaria-endemic area are subject to a plasma-only restriction period of at least 12 months. The restriction period is extended to 3 years if residence has been for 6 continuous months or more within the past 3 years. With a negative malaria test, the restriction period can be reduced to 4 months. Donors who have traveled to an HIV risk area must be asked if they had sexual contact with a resident of that area. | |||||
Appendix 3: Disease Testing
| Disease Test | United States | European Union | ||
|---|---|---|---|---|
| Hepatitis | ||||
| Permanent deferral for repeatedly reactive test results (FDA) | European Pharmacopoeia:
Human Plasma for Fractionation |
|||
| Permanent deferral if reactive on 2 or more separate occasions; permanent deferral if core antibody positive results are coupled with prior or concurrent repeatedly reactive HBsAg test (FDA). Testing is not required for Source Plasma donors | Laboratory tests are carried out for each donation to detect the following viral markers: (1) antibodies against HIV-1 (anti-HIV-1). (2) antibodies against HIV-2 (anti-HIV-2). (3) hepatitis B surface antigen (HBsAg). (4) antibodies against hepatitis C virus (anti-HCV). If a repeat-reactive result is found in any of these tests, the donation is not accepted. |
|||
| Permanent deferral if repeatedly reactive for hepatitis C antibody; may re-enter donor after 6 months if confirmatory test is negative (FDA) | Blood Directive 2002/98/EC (Annex IV): Basic Testing Requirements for Whole Blood and Plasma Donations | |||
| Permanent deferral if positive on single testing (FDA) | The following tests must be performed for whole blood and apheresis donations, including autologous predeposit donations: | |||
| ABO group (not required for plasma intended only for fractionation). Rh D group (not required for plasma intended only for fractionation). Testing for the following infections is required in donors: Hepatitis B (HBsAg), Hepatitis C (Anti-HCV), HIV 1 and 2 (Anti-HIV 1 and 2). Additional tests may be required for specific components or donors or epidemiological situations. Blood Directive 2004/33/EC (Annex III): Permanent Deferral Criteria (excerpt): Hepatitis B, except for HBsAg-negative persons who are demonstrated to be immune Hepatitis C HIV 1 or 2 |
||||
| HIV | ||||
| Permanent deferral if positive on single testing (FDA) | ||||
Antibody (EIA) |
Permanent deferral for repeatedly reactive HIV EIA test; may reenter after 6 months if confirmatory test negative (FDA). Testing is not required for Source Plasma donors | European Pharmacopoeia: Human Plasma for Fractionation
Laboratory tests are carried out for each donation to detect the following viral markers: (1) antibodies against HIV-1 (anti-HIV-1) (2) antibodies against HIV-2 (anti-HIV-2) (3) hepatitis B surface antigen (HBsAg) (4) antibodies against hepatitis C virus (anti-HCV) If a repeat-reactive result is found in any of these tests, the donation is not accepted Blood Directive 2002/98/EC (Annex IV): Basic Testing Requirements for Whole Blood and Plasma Donations The following tests must be performed for whole blood and apheresis donations, including autologous predeposit donations: ABO group (not required for plasma intended only for fractionation) Rh D group (not required for plasma intended only for fractionation) Testing for the following infections are required in donors: Hepatitis B (HBsAg) Hepatitis C (Anti-HCV) HIV 1 or 2 (Anti-HIV 1 and 2) Additional tests may be required for specific components or donors or epidemiological situations. Blood Directive 2004/33/EC (Annex III), Permanent Deferral Criteria (excerpt) Hepatitis B, except for HBsAg-negative persons who are demonstrated to be immune Hepatitis C HIV 1 or 2 |
1
FDA. Blood Guidances. www.fda.gov/cber/blood/bldguid.htm. FDA Memoranda to Blood Establishments available at www.fda.gov/cber/memo.htm.
2
PPTA. www.pptaglobal.org.
3
AABB. Standards for Blood Banks and Transfusion Services. 25th ed. Bethesda, MD: AABB; 2008.
4
FDA. Guidance for Industry: Quality Systems Approach to Pharmaceutical cGMP Regulations. 2006. Available at www.fda.gov/cber/gdlns/qualsystem.htm.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Anita Y. Szajek, Ph.D.
Principal Scientific Liaison 1-301-816-8325 |
(GCBA2010) General Chapters - Biological Analysis |
USP35–NF30 Page 809
Pharmacopeial Forum: Volume No. 35(2) Page 388