INTRODUCTION
The basic principles of nucleic acid amplification technologies (NAT) and definitions of the various techniques are covered in Nucleic Acid–Based Techniques—General  1125
1125 . The current chapter covers major techniques that result in amplification of targeted nucleic acid sequences. The most common NAT assay is the polymerase chain reaction (PCR), which was first described by Kary Mullis. This procedure has been further refined to amplify a DNA fragment starting from RNA (reverse transcription-PCR, or RT-PCR). Initially, PCR was used in a qualitative manner to amplify and detect DNA molecules because its exquisite sensitivity paired with its high specificity made it a useful tool for the detection of nucleic acid targets. Since its inception, the number of PCR applications has expanded rapidly, and the technique, which now includes quantitative and multiplex assays, is currently used in almost every field of research and development in biology and medicine. In addition to the changes and improvements to the original design of the PCR procedure, alternatives to PCR are techniques used to amplify target nucleic acids to generate RNA instead of DNA amplicons. The most commonly used techniques are nucleic acid sequence–based amplification (NASBA) and the transcription-mediated amplification (TMA) which are described here in detail. In contrast to PCR, which relies on incubating the sample at three different temperatures, NASBA and TMA are based on isothermal conditions.
. The current chapter covers major techniques that result in amplification of targeted nucleic acid sequences. The most common NAT assay is the polymerase chain reaction (PCR), which was first described by Kary Mullis. This procedure has been further refined to amplify a DNA fragment starting from RNA (reverse transcription-PCR, or RT-PCR). Initially, PCR was used in a qualitative manner to amplify and detect DNA molecules because its exquisite sensitivity paired with its high specificity made it a useful tool for the detection of nucleic acid targets. Since its inception, the number of PCR applications has expanded rapidly, and the technique, which now includes quantitative and multiplex assays, is currently used in almost every field of research and development in biology and medicine. In addition to the changes and improvements to the original design of the PCR procedure, alternatives to PCR are techniques used to amplify target nucleic acids to generate RNA instead of DNA amplicons. The most commonly used techniques are nucleic acid sequence–based amplification (NASBA) and the transcription-mediated amplification (TMA) which are described here in detail. In contrast to PCR, which relies on incubating the sample at three different temperatures, NASBA and TMA are based on isothermal conditions.
In addition to amplification of the target nucleic acid, the amplification step also can be directed at the signal used for detection (signal amplification). The most commonly employed technique is the branched DNA (bDNA) assay, in which the signal, typically a fluorescent probe that binds to the target sequence, is amplified. The bDNA assay is used predominantly for viral nucleic acid detection and quantitation.
This chapter describes the main assay components necessary for a PCR procedure and includes a discussion of the general optimization of PCR assays. The various PCR assay formats, including PCR, nested PCR, and RT-PCR are covered, and a discussion of the detection of the resulting amplicons follows. Although all these assays are essentially qualitative procedures, they can be modified for semiquantitation, and the various modifications are described. For accurate and reliable quantitation, real-time PCR has now replaced the methods listed above; real-time PCR and real-time RT-PCR are described in the NAT Assays section. The same section includes a discussion about probes and dyes that are an essential component of real-time PCR and the methods of quantitation using the generation of standard curves. The next PCR technique discussed is multiplex PCR, which is used for simultaneous detection of multiple targets or for normalization of assay results. Apart from PCR, the major alternative NAT tests that are used routinely, primarily in blood screening and clinical diagnostic screening are NASBA and TMA. The final technique described is whole-genome amplification, wherein the complexities of amplification require modifications to the PCR procedures. The chapter concludes with a discussion about the evolution of instrumentation used in NAT assays and the quality assurance and quality control issues associated with NAT because this is probably one of the most highly regulated biological techniques, especially when applied to blood screening.
ASSAY COMPONENTS
Enzymes
The essential components for NAT assays—polymerases, reaction buffers which include desoxynucleotides, ions, primers, probes, and fluorescent dyes—can be chosen from a broad selection of commercially available NAT reagent kits and vendors. Polymerases suitable for NAT applications can, in principle, be grouped into Taq DNA polymerases or DNA I polymerases from other Thermus species that are polymerases with features that are similar to those of Taq DNA polymerase. In addition, so-called proofreading polymerases are available (e.g., from Pyrococcus species) that display a 3¢–5¢ exonuclease activity capable of removing wrongly incorporated DNA bases from the growing DNA strand under amplification conditions. Taq DNA polymerase is the standard NAT enzyme and is the one most often used in NAT assays. Modifications of Taq DNA polymerase, such as deletions of the 5¢–3¢ exonuclease domain (Klenow fragment, Stoffel fragment) or point mutations for improved incorporation of dideoxynucleotides are also employed (e.g., for PCR-based sequencing reactions). Proofreading DNA polymerases or mixtures of Taq DNA polymerase with a proofreading polymerase are used if either fidelity of the NAT product is critical (e.g., for DNA cloning experiments) or longer NAT products are to be amplified. For RT–PCR, a reverse transcriptase is necessary to first convert the RNA target to copy DNA (cDNA) that can subsequently be amplified. For TMA reverse transcriptase with an RNAse H activity is needed to convert the RNA target to double-stranded template DNA, while for NASBA exogenous RHAse H is added to the reaction mixture. Depending on the reaction environment, two types of enzymes can be used to generate cDNA: a reverse transcriptase isolated from retroviral sources or a DNA polymerase that can function both as reverse transcriptase and DNA polymerase. Finally, chemical modification of the polymerase, resulting in an inactive enzyme at temperatures below 90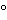 , is now typically used to prevent mispriming of templates at sub-optimal temperatures (see section on Assay Optimization).
, is now typically used to prevent mispriming of templates at sub-optimal temperatures (see section on Assay Optimization).
Reaction Buffers
Reaction buffers vary with respect to ion composition, pH, and additives and are sometimes specifically adopted for particular applications such as multiplex PCR, real-time PCR, RT–PCR, TMA and NASBA. An important component of the reaction mixture is Mg2+ ions, or, in the case of polymerases with both reverse transcriptase and DNA polymerase functions, such as Thermus thermophilus (Tth), Mn2+ ions. Other additives that enhance the sensitivity and specificity of the assay may be present. The concentration of the four deoxynucleotide triphosphates (dNTPs) must be optimized.
Primers
Primer sets are oligonucleotides with sequences that are designed specifically to prime the amplification of a portion of a target nucleic acid of interest. Synthetic oligonucleotide primers for both standard PCR and for real-time or quantitative PCR are designed for the specific recognition of and binding to a single DNA or RNA sequence. Such specificity is achieved through design that involves both the length and the sequence of the primers. Length and sequence specifications have separate criteria that must be simultaneously met in order for the primers to perform properly. The length of a primer is a statistical issue that relates to the issue of the minimum length of a specific sequence necessary to guarantee that the desired target sequence is unique, regardless of the size or complexity of the genome. As an example, in the case of the human genome, with its 3.2 billion DNA bases, that length is 17 bases. For this reason the vast majority of PCR primers are between 20 and 25 bases long. The specificity of a primer should be determined by comparison with sequences in all known databases. Tools available on the Web facilitate such comparisons.
In terms of primer sequence, the issues to consider are Tm (the temperature at which 50% of the double-stranded nucleic acid molecule becomes single-stranded) and secondary structure. Every DNA has its own characteristic Tm, determined by length, sequence composition, and reaction environment. PCR primers are designed to bind to a perfectly complementary DNA sequence via guanine-cytosine (G-C) and adenine-thymine (A-T) base pairing. The Tm of the two PCR primers used in a reaction should be as close as possible. In terms of secondary structure, the formation of secondary structures by intra- or intercomplementarity should be minimized. Interaction between different primers can result in primer–dimers that will compromise assay sensitivity and specificity. All of the design issues presented are accounted for in any one of the dozens of primer design software packages that are available and can be found on the Internet.
Assay Optimization
NAT assay optimization is necessary for successful amplification that is sensitive and specific. Parameters that should be optimized include the thermocycling conditions, both temperatures and cycling times (that depend to a large extent on the target, primer, and probe sequences), concentrations of template, concentrations of NAT reagents, sample matrix and the number of amplification cycles. In the case of multiplex PCR, a compromise among elements of the reaction conditions is usually necessary because of the difficulties in optimizing the conditions for all the primer and probe sets. Recent changes have been made to improve sensitivity and specificity of NAT assays. One change is hot-start PCR, in which the addition of one of the essential components of the NAT assay, typically the DNA polymerase, is temporarily withheld. When this occurs during reaction setup, the initial nucleic acid denaturation step prevents nonspecific amplification due to mispriming at suboptimal temperatures. Early hot-start procedures made use of wax barriers that effectively separated essential components into two liquid phases that were mixed only when the wax melted. However, this procedure has been replaced by two important hot-start technologies that do not require physical separation of the components by inconvenient additional handling steps. In the first procedure, antibodies directed against the DNA polymerase are complexed with the enzyme and lose their binding avidity at elevated temperature at the start of the reaction. The second procedure uses chemical modification of the polymerase, resulting in an inactive enzyme. At temperatures above 90, typically in the first denaturation step, the modifier dissociates from the enzyme, and the enzymatic activity is restored. The advantage of an antibody-mediated hot start is the immediate release of enzyme activity at the start of the reaction by a very short heat incubation step. However, antibody-mediated hot-start chemistries tend to be less stringent when compared with chemically activated enzymes if there is a large excess of active polymerase molecules.
NAT ASSAYS
This section describes the basic techniques of PCR, nested PCR, and RT-PCR and procedural modifications that allow semiquantitation.
Polymerase Chain Reaction
The PCR technique is based on a three-step process: denaturing double-stranded DNA (dsDNA) into single strands (ssDNA), annealing primers to the ssDNA, and enzymatic extension of primers that are complementary to the ssDNA templates. Each step is usually carried out at a different temperature. By cycling the temperature steps many times (usually 30 to 45 times), a billion-fold amplification of the target nucleic acid can be achieved, but the optimal number of cycles should be determined empirically. In some cases, especially where sensitivity is more important than false positive results due to excessive cycling, such as in blood screening, extra sensitivity can be gained by increasing the number of cycles to 60 to ensure that extremely low levels of target are detected. In a typical reaction, PCR product (amplicon) doubles at each cycle of amplification (exponential amplification). The increase in amplification in the early cycles follows a sigmoidal curve. In later cycles, the concentrations of the template strands and amplicons favor template strands re-annealing instead of PCR primer annealing to the template. At this point the concentration of the PCR product no longer doubles after each cycle, and the curve begins to plateau. A thermostable enzyme such as Taq-polymerase is a prerequisite because temperature cycling at 95 (the typical temperature step used to denature double-stranded templates) would inactivate a thermolabile polymerase.
Nested PCR
An early variation of the PCR assay was nested PCR, which was designed to increase the assay's sensitivity and specificity. In this procedure amplicons from the initial PCR reaction are subjected to a second round of amplification using a different set of primers. This set of primers is specific to the amplicon sequence but is within the first set of primers (nested primers). The advantage of amplification with two sets of target-specific primers is increased specificity (any nonspecific amplification during the first amplification round would be reduced) and increased sensitivity (due to initial amplification of the target in the first amplification round). In addition, amplification of a product of the expected size is taken as confirmation of the presence of the target. However, a major drawback of this procedure is the high likelihood of cross-contamination due to the increased manipulation of amplicons generated in the first round of amplification. The use of highly specific primers and probes and the optimization of reaction conditions have resulted in the diminished applications of this procedure for routine testing, but the procedure is sometimes used for samples that are difficult to amplify by conventional PCR.
RT-PCR
In amplifying RNA targets, analysts prepare cDNA before the amplification step (RT-PCR). One-step and two-step RT-PCR procedures are available. In one-step RT-PCR the reverse transcription of RNA into cDNA and the subsequent amplification step are carried out in a single reaction without intermediate procedures. Therefore the reaction mixture for one-step RT-PCR includes the gene-specific amplification primers that are used for both reverse transcription and amplification. The advantage of this procedure is the overall reduction in handling time, increased throughput, and reduced contamination risk because reopening the reaction vessel is not necessary. In contrast, in two-step RT-PCR the reverse transcription and amplification are performed as two separate steps. In general, random primers or oligo-d(T) primer rather than gene-specific primers are used for the reverse transcription step. An aliquot of the cDNA synthesis reaction is then transferred into the NAT reaction for subsequent amplification. The advantage of this procedure is the standardization of the reverse transcription reaction, which can be used as a single source for the analysis of multiple transcripts in gene expression analysis.
Detection of Amplicons
Following amplification, analysts can employ a variety of procedures for detection of the amplicon as described in detail in the general information chapter, Nucleic Acid–Based Techniques—Extraction, Detection, and Sequencing 1126. These include agarose gel electrophoresis with ethidium bromide or other dyes, capillary electrophoresis, and laser-induced fluorescence and hybridization followed by chromogenic detection such as streptavidin horseradish peroxidase detection, chemiluminescence, or fluorescent detection using labeled probes.
Quantitation—
The original PCR and RT-PCR assays were qualitative and detected amplicons at the end of the reaction. Such detection is not easy to quantitate because at this stage the amplification is in a plateau phase at the end of the assay, and the amount of amplicon is not necessarily directly related to the quantity of the starting template. Several approaches have been deployed to attempt to overcome the shortcoming of PCR to produce reliable, quantitative results. Initial attempts at quantitation relied on assessing the amount of amplified DNA during the early or exponential part of the assay, but this procedure was fraught with problems because the aliquots had to be taken from the reaction mixtures at regular intervals, thus greatly increasing the risk of cross-contamination. One of the earliest and most straightforward approaches to quantifying PCR products was to measure the amount of amplicons that were generated during the exponential phase of the reaction by comparing this to a serially diluted external control. Several aspects, including variability in sample preparation and variations in reaction conditions, however, hampered this approach. Because of the exponential amplification of NAT procedures, even small errors or variances can lead to distinct differences.
Compared with dilution procedures, competitive PCR proved to be a much more precise approach to achieving reliable estimates of the originally present target molecules. This procedure relies on the simultaneous co-amplification of a specific target sequence in the presence of increasing concentrations of an exogenous target molecule (control) which shares the primer binding sites with the target sequence but whose sequence is slightly modified or shortened in order to facilitate discrimination from wild-type amplicons. In addition, the concentration of the control is known. The close sequence homology and similar size of the control and target amplicons are designed to ensure that the template and internal control are amplified with comparable efficiency. The relative strength of the amplicon bands of template and control can be assayed, for example, on ethidium bromide–stained agarose gels, giving a relatively precise quantitation of the wild-type target. A drawback of this approach is that the internal control and the template should be present in the reaction in approximately the same quantity in order to yield correct results. The development of real-time, quantitative PCR has eliminated the variability associated with quantitative PCR, thus allowing the routine and reliable quantification of PCR products.
Real-Time PCR and Real-Time RT-PCR
Although gene quantitation by quantitative PCR was a widely used procedure, its applications were expanded by the advent of real-time PCR and real-time RT-PCR. Real-time PCR displays the same advantages as standard quantitative PCR—sensitivity, specificity, and a wide dynamic range—but the real-time procedure offers the additional advantage of requiring no post amplification processing because it combines amplification and detection in a single step. Real-time PCR collects data throughout the amplification process by measuring a fluorescence signal created as amplification progresses. A multitude of fluorescence chemistries allows the correlation of generated PCR product to fluorescence intensity. In principle, fluorescence intensity will increase with every cycle performed. Once the intensity is greater than background fluorescence, the so-called cycle threshold (Ct) value is achieved. This value, which represents the first cycle in which there is a detectable increase in fluorescence above the background level, is used to measure relative or absolute target quantities. The Ct value is inversely proportional to the number of target molecules in the sample and thus provides a means to quantitate the amount of target in the starting material (i.e., the greater the number of target molecules present, the lower the Ct value).
The reaction conditions for real-time PCR applications have to take into account the presence of the probe(s) and will require optimization. The most commonly used probes currently are hydrolysis probes, although hybridization probes are an alternative. In most cases, the amplification and detection steps can be combined into a two-step cycling reaction, but these conditions have to be optimized. In contrast, DNA-binding dyes which may also be used for amplicon detection require separation of the annealing and extension steps since the dye binding occurs during the extension step which is usually done at 72.
A fluorescent DNA intercalating dye is used for detection of the PCR product in real-time mode. This dye emits light when bound to double-stranded DNA and the subsequent increase in fluorescence can be detected by real-time PCR instruments. Dyes that bind to dsDNA bind not only to the specific PCR product but also to artifacts such as nonspecific PCR products and primer–dimers. Analysts have observed substantial differences in the specificity of dsDNA-binding dyes in use with real-time PCR kits. Therefore, some analysts recommend verifying the presence of a single PCR product by gel electrophoresis to determine the correct size of the PCR product. Also, a melting curve analysis is advisable to ensure the absence of artifacts that could contribute to the fluorescent signal and thereby lead to misinterpretation of quantitative data. Alternatively, sequence-specific labeled probes can be employed. A wide variety of fluorescence-labeled probes and primers exist for use in real-time PCR and are described in the next section.
Real-Time PCR Probes—
The difference between conventional PCR and real-time PCR is the presence of a third chemically synthesized oligonucleotide, the probe, which, for the most basic hybridization probes, contains some type of reporter molecule, usually a fluorescent molecule or fluorophore. Non-nucleic acid materials can be added to chemically synthesized DNAs that are then incorporated into oligonucleotide probes for real-time PCR. Other applications include hybridization probes such as those used for fluorescence in situ hybridization (FISH) and microarrays and probes designed to capture other nucleic acids. A challenge arises in using fluorescent probes for real-time PCR because the unbound or free probe is not removed before detection, thus requiring a means to distinguish between signal obtained from bound and free probe. In contrast, FISH assays involve washing away free probe following hybridization.
All of the issues associated with primer design for conventional PCR apply to real-time PCR primers as well as to the probe sequence. As a general rule only two additional considerations apply to the probe sequence. One of these is thermodynamic, and the other specifically concerns the reporter moieties themselves. Thermodynamically, a good probe molecule that is designed to bind in the sequence somewhere between the two PCR primers will have a Tm that is about 5 higher than that of the two primers. In the large majority of cases the amplicon will be between 100 and 500 DNA bases in length, although for real-time PCR a smaller amplicon between 100 and 150 bases long results in a more efficient reaction. Thus it is rarely a problem to find a sequence inside a PCR amplicon that meets the necessary criteria.
Current probe designs overcome the problems of background from unbound probe using simple hybridization probes. In the original design, two probes that hybridize to adjacent sequences on the target nucleic acid are labeled. The reporter moiety is a fluorescent molecule attached to the 3¢ end of the upstream probe sequence, and a second fluorescent molecule is attached to the 5¢ end of the second probe. Excitation of the 5¢ fluorophore with light energy of the proper wavelength results in absorption of that energy, followed by emission of light energy of a slightly longer or less energetic wavelength (Stoke's Law). This emitted energy then excites the 3¢ fluorophore if it is close enough to the emitter and compatible with it in the sense that the emitted energy from the 5¢ fluorophore can excite the 3¢ fluorophore. When this occurs, the observed fluorescent light wavelength will be that of the acceptor molecule and not that of the donor. Fluorescence absorption and emission spectra are readily available for all of the commonly used fluorophores, and the only applicable rules are that the two fluorescent molecules must be fewer than 40 DNA bases apart and that the emission spectrum of the donor must overlap the absorption spectrum of the acceptor. Thus hybridization of the two probes, also known as hybridization probes or FRET probes (Fluorescence Resonance Energy Transfer), results in the emission of a fluorescent signal by the acceptor, and the latter signal can be detected. In the absence of hybridization, the probes are sufficiently separated in solution so that energy transfer cannot occur, and only background fluorescence is emitted by the donor.
Issues of fluorophore compatibility have been resolved by the increased use of a special class of molecule called a quencher. Quenchers are fluorescent molecules that absorb fluorescence energy over a wide range of wavelengths. Instead of re-emitting that energy as light they simply dissipate it as heat. Thus, if a quencher molecule is placed at the 3¢ end of a probe and a fluorophore at the 5¢ end, the probe will remain dark even when excitation energy is present so long as the molecule remains intact (hydrolysis probes). These probes utilize the 5¢ nuclease activity of the DNA polymerase to hydrolyze a probe bound to its target amplicon. Cleavage results in separation of the reporter and quencher and permits fluorescence of the reporter. This reduces much of the work of optimization of the assay conditions (since only a single probe is used) and background noise generated with two probes.
A variation on hydrolysis probes involves placing the reporter and quencher molecules on a single oligonucleotide that is constructed so that, in the unbound state, the quencher and reporter are in close proximity, resulting in efficient quenching of the reporter. When the probe hybridizes to its complementary sequence on the amplicon, the probe undergoes a conformational change that forces the quencher and reporter apart, permitting fluorescence of the reporter. A variation on these kinds of probes is a combined primer and probe in which, again, the quencher and reporter are in close proximity in the native probe, thus resulting in no signal. Priming and subsequent elongation of the primer–probe results in hybridization to the newly synthesized DNA strand, causing spatial separation of the quencher and reporter and resulting in the generation of a signal.
Probe Labeling—
Modern synthetic oligonucleotide modification chemistries permit the manufacture of oligonucleotides with non-nucleic acid materials. Placement of modifications is carried out in one of two ways: during synthesis or after synthesis. For the former, modifications are constructed in such a way that they behave like the four DNA or RNA bases that are routinely placed in the sequence. The modification is then presented in the desired location during the synthesis as if it were just another base in the series. In the latter, usually employed when more than one modification occurs, the synthesis contains a linker, such as an amino group, to which the desired modification is then attached. This process is often called “hand-tagging.”
Perhaps the best-known example of hand-tagging is the conventional dual-labeled probe used in real-time PCR. The quencher is placed at the 3¢ end of the sequence during synthesis, and the fluorescent reporter molecule is hand-tagged to an amino modification at the 5¢ end of the sequence after the synthesis is finished and has undergone purification. Some modifications, such as biotins, are designed so that multiple modifications can be carried out in a single synthesis. Thus, it is possible to modify a synthetic DNA or RNA sequence to contain a number of different non-nucleic acid molecules. A cost is associated with such modifications insofar as alterations often are achieved with a loss of mass due either to an inherently lower efficiency of modifications to bind to the oligonucleotide as compared with standard DNA or RNA bases or to the requirement that the synthesis must be purified before modification, after modification, or both.
The benefits of modifying synthetic DNAs or RNAs usually outweigh the costs. The standard, quenched, dual-labeled, real-time PCR probe has permitted precise quantification of gene expression. Fluorescently labeled DNA oligonucleotides are also essential components of in situ hybridizations and microarrays. Some modifications confer increased thermal stability when synthetic DNAs or RNAs are hybridized to complementary DNAs or RNAs by comparison with unmodified DNA–DNA and DNA–RNA duplexes. These analogues include peptide nucleic acids, 2¢-fluoro N3-P5¢-phosphoramidates, and 1¢, 5¢-anhydrohexitol nucleic acids. Although such analogues succeed to varying degrees in achieving increased thermal stabilities, they fail to provide enhanced target recognition. Another approach is to use base analogues such as locked nucleic acid, which is an analogue that contains a 2¢-O, 4¢-C methylene bridge. This bridge restricts the flexibility of the ribofuranose ring and locks the structure into a rigid bicyclic formation, conferring enhanced hybridization performance and stability.
The modification of a probe typically is governed by its intended use. Generally, fluorescent reporters are used in real-time PCR and for in situ hybridization. The range of available fluorescent reporters covers the spectrum from 517 nm to 778 nm. For hybridization probes, base modifications are preferred because these primarily alter thermodynamic interactions between bases, leading to improved specificity. Amino attachment groups, both with and without C-spacers, are used to attach other modifications to DNA sequences and to attach DNA sequences to solid surfaces such as glass slides. An example is the attachment of biotin molecules to DNA sequences. Biotin forms a strong bond with streptavidin-coated materials such as magnetic beads, allowing capture of specific nucleic acids that may themselves be hybridized to other molecules.
Quantitation—
PCR products may be quantified using a standard curve drawn from replicate serial dilutions of a reference reagent or standard for the nucleic acid sequence of interest. The concentration of the nucleic acid in the reference reagent is known. Real-time PCR quantitation based on a standard curve may utilize plasmid DNA or other forms of DNA. However, the efficiency of PCR must be the same for the standards and the target samples. Performing PCR from purified targets can in some cases be more efficient than performing PCR with complex nucleic acid mixtures. The cycle threshold (Ct) values and concentrations of the dilutions of the reference reagent can be used to construct a standard curve from which the concentration of the unknown sample can be estimated. When the assay run conditions have been well standardized and the standard curve for a particular target has been well calibrated, in subsequent assay runs it may be sufficient to co-amplify only two dilutions of a reference reagent (usually dilutions containing known amounts of nucleic acid at high and low concentrations). These dilutions, or calibrators, can then be used to quantitate any unknown samples by comparison of the Ct values.
Multiplex PCR—
Multiplex PCR describes the simultaneous amplification of several nucleic acid targets in a single assay reaction. This is a particularly demanding variation of PCR because it requires the use of a single set of reaction conditions for the amplification of multiple targets with different sequence characteristics. Additional complications can arise due to the increased chance of nonspecific amplification products arising from multiple primer interactions. In addition, the differing individual target amplification efficiencies can result in weaker reactions being out-competed by stronger, more efficient reactions.
Both qualitative and quantitative applications of multiplex PCR have been described in the literature, as have multiplex RT-PCR assays. Quantitative multiplex PCR relies on either the generation of multiple standard curves to enable quantitation of each target in the assay, or the inclusion of internal competitor sequences that can be used as calibrants.
Hybridization kinetics of primers and probes may be significantly different, even when designed using the same algorithm. This leaves the analyst with very limited room to optimize reaction conditions. However, optimization may include adjustment of DNA polymerase amount, Mg2+ to increase hybridization efficiency, or primer concentration. Especially in real-time PCR, optimization of primer concentration is critical for quantitative co-amplification of target genes. These are contained in the sample at significantly different amounts. Increasing hybridization efficiency of the primer–probe system can be achieved by providing sufficient reagents, such as Mg2+, as well as adding a “molecular crowding” reagent that increases the effective concentration of all reaction components in the mixture. Multiplex PCR is not only used for genotyping applications, but also for quantitative real-time PCR because it offers several advantages over standard single real-time PCR reactions. Some of these advantages are a minimized amount of sample used, increased precision through the use of an internal control (e.g., housekeeping gene) co-amplified with the target gene in the same reaction, no separate pipetting steps, and cost-effectiveness.
Most PCR assays, however, suffer from a common problem—that of minimizing differences in extractions or amplifications between different samples. Multiplex PCR is useful in cases where it is critical to ensure that variability in quantitation of different samples is not due to differences in nucleic extraction or amplification measurements (usually when one measures the production of an mRNA species). Certain precautions and techniques can be employed to minimize these challenges; they are discussed in the next section on normalization of assay results.
Normalization of Assay Results—
To minimize the effects of assay variables, analysts sometimes use a relative quantitation procedure that normalizes the target transcript level to a control that can be employed and compared for all samples included in the gene expression study. Probably the most reliable and most frequently used relative quantitation procedure relies on the measurement of “housekeeping” or control genes to normalize the expression of the target gene in a multiplex PCR format. This procedure is preferred because the quantitation of both the housekeeping gene and the target gene are influenced by varying cDNA synthesis efficiencies or the presence of enzyme inhibitors contained in the sample. However, it should be noted that the efficiency of conversion of target RNA to cDNA is not necessarily consistent even within a single-tube reaction but is a function of primer design, target sequence, etc. which may differ between target and housekeeping genes. The selection of appropriate control genes can cause problems because they may not necessarily be equally expressed across all unknown samples and may vary under experimental conditions. Normalizing measurements to a set of housekeeping genes in order to avoid the problem of variability may circumvent this concern. Alternatively, analysts can establish a thorough evaluation of housekeeping genes that do not alter gene expression levels under the experimental conditions.
All the NAT techniques described thus far are variations on the PCR assay, which is the most widely used of the NAT techniques. However, isothermal assays that are based primarily on the amplification of RNA are used for routine purposes. This is known as the transcription-mediated amplification (TMA) assay, which is closely related to the nucleic acid sequence–based amplification (NASBA) assay. Both assays are described in more detail in the following section.
Nucleic Acid Sequence-Based Amplification and Transcription-Mediated Amplification
Both NASBA and TMA rely on in vitro isothermal amplification for detection and amplification of nucleic acids, also referred to as self-sustained sequence replication or 3SR. The major difference between the assays is that NASBA uses three enzymes—reverse transcriptase (RT), RNA polymerase, and RNase H—whereas TMA uses only two enzymes: RT and RNA polymerase. The complete procedure generally is performed at 41 to 42 using two primers. Both NASBA and TMA are especially suited to amplifying RNA analytes, including rRNA, mRNA, pathogens that have RNA as their genetic material, as well as DNA targets.
One of the primers that has a promoter sequence for the RNA polymerase at the 5¢ end binds to the RNA target and is extended via the DNA polymerase activity of the RT. The product of this reaction is an RNA–DNA hybrid. RNase H activity then specifically digests the RNA strand of the hybrid, leaving only the cDNA to which the second primer can bind. A complementary strand of DNA is then synthesized by the RT, resulting in a dsDNA molecule with a T7 promoter at the 5¢ end. The T7 RNA polymerase then transcribes multiple copies of the RNA amplicon. The RNA copies may undergo the same cycle to create new duplex DNA molecules with a T7 promoter from which many molecules of RNA are transcribed. Thus, unlike the action of PCR, the amplicon amplified in this case is of an RNA species.
Some of the characteristics of this technology are that only relatively short target sequences can be amplified efficiently (around 100–250 nucleotides); it uses a single temperature, which eliminates the need for special thermocycling equipment; the fidelity of the technique is comparable to that of other amplification processes; and the RNA amplicons are exponentially amplified. Carryover contamination is minimized because of the labile nature of the RNA amplicon in the laboratory environment. Containment procedures built into the assay procedure further help to minimize contamination. Detection of amplicons is typically achieved by the use of labeled probes and, in TMA technology, a common method is detection of chemiluminescent signals from hybridized probes that remain intact during the subsequent alkaline hydrolysis step used to destroy free probe.
The NAT techniques described, both PCR and TMA, are optimized for amplifying specific, small fragments of a genome. In cases when whole genome amplification is desirable, such as for mutation analysis or identity testing, modifications of the PCR procedure are necessary in order to ensure adequate sequence representation of genetic loci, as described in the following section.
Whole Genome Amplification
Historically, whole genome amplification (WGA) has been performed using modified PCR procedures. These procedures have relied on the nonspecific amplification of the genome using primers that bind under low-stringency conditions to the DNA template. PCR-based approaches differ mainly in terms of the type of primer employed in the reaction: in primer-extension-preamplification (PEP), short 15 base random primers are used in an initial cycling reaction at low stringency to make multiple random copies of segments of the genome. This product is then used as target for the specific PCR reaction. Amplification bias of favorable sequence contexts leading to uneven representation of the genome is the major drawback of this technique. The generation of increasingly shorter fragments during each round of amplification is a further drawback. Another procedure called degenerate oligonucleotide primed-PCR (DOP-PCR) uses tagged primers and low stringency amplification for the first few cycles of amplification followed by an increase in annealing stringency in later cycles. The tagged primers are characterized by defined sequence tags at the 3¢ and 5¢ ends and a random sequence in the centre of the primer. Under the later, more stringent conditions, the target DNA fragments generated during the first cycles containing the amplification tag sequences are amplified preferentially without any further shortening of the fragment length. PCR-based WGA typically employs Taq-like polymerases that possess the disadvantage of introducing variations into the amplified DNA due to their relatively low processivity and fidelity which become compounded by the very high number of amplification cycles used in these methods. This may cause problems in downstream applications such as genotyping analysis. These limitations as well as the relatively poor sequence representation of genomic loci inherent to PCR-based WGA can be overcome by an isothermal reaction called multiple displacement amplification (MDA).
The enzyme that is used for MDA comprises a high processivity polymerase with proofreading and strand-displacement activity. The isothermal reaction is performed at 30 without any change in reaction temperature. The reaction starts with the annealing of multiple random primers to the target DNA and elongation of the primers using a DNA polymerase from the Bacillus subtilis phage Phi29. Because the polymerase is able to displace DNA strands in a 5¢–3¢ direction, the polymerase reaction is not stopped when the elongating strands meet downstream DNA strands. The displaced DNA strand serves again as a target for multiple primed elongation reactions so that the DNA template is amplified exponentially in a branched-like manner, yielding high molecular weight DNA with a good representation of the genomic loci. Compared with PCR-based WGA, the error rate is very low. In particular, the mutation rate of repetitive sequence structures is low because of the limited strand-displacement activity of Phi29-polymerase. This permits reliable genotyping of genomic DNA (e.g., SNP analysis, mutation analysis, identity testing, or analysis of case work samples) on different platforms such as real-time PCR or array analysis.
INSTRUMENTATION
The development of the numerous and varied NAT techniques described in this chapter has been facilitated by the evolution of instrumentation that has served to automate these complex procedures. A general description of the major changes in instrumentation is discussed in this section.
The continuous control of the temperature steps necessary to achieve exponential amplification for PCR assays is carried out by fully automated thermocyclers that consist of a heating block in which the temperature can be rapidly cycled. Temperature changes are induced by water, or more recently, by using the Peltier effect. These instruments may be coupled to a fluorometer apparatus if they are used for real-time PCR analysis. In the latter case certain instruments are equipped with a rotor device that is heated and cooled by air instead of a metal block that typically is used as a heating module. In the case of endpoint PCR, PCR products are usually analyzed according to size on agarose or polyacrylamide gels, or by capillary electrophoresis using fluorophore-labeled primers. They may also be analyzed by an array-based approach or other hybridization procedures.
Because no post-PCR processing or label-separation steps are required, real-time PCR assays are simple to perform, making them useful for high-throughput applications. Real-time PCR instruments combine the properties of a thermocycler and a fluorometer to allow determination of PCR products by fluorescence measurement. In each PCR cycle, either one or several fluorescence readouts are taken to monitor the PCR reaction for generation of amplicons, usually at the extension step of the PCR reaction.
Real-time PCR instruments vary with regard to simultaneous sample throughput (32–384 reaction vessels), sample volume (5–100 µL), excitation source, and detector used. These compositions define the suitable range of fluorescent dyes for multiplex real-time PCR as well as size and heating/cooling principle (see above). The excitation source of real-time thermocyclers is either a laser-based system, halogen bulbs, or light-emitting diodes (LED). Optical filters are used to select the wavelength of interest. In most instruments, the emitted light is detected by a charge-coupled device (CCD) that consists of an array of light-sensitive cells. Light projected onto the CCD is converted to an electric charge, resulting in a signal that is proportional to the light intensity.
The versatility of the PCR assay has resulted in the widespread and diverse use of this technique. With the advent of real-time PCR, it has been possible to design high-throughput instrumentation for automated testing. Similarly, the TMA assay has also been automated. Such technology is used by laboratories doing high-throughput, highly regulated testing, typically blood screening for hepatitis C virus (HCV) or human immunodeficiency virus-1 (HIV-1) because automated tests are ideal in a regulated environment where minimum human intervention is required. The use of NAT in a highly regulated environment has resulted in the development of guidances for managing the quality assurance (QA) and quality control (QC) aspects of testing, as well as the validation of systems and assays as described in the following section.
QUALITY ASSURANCE AND QUALITY CONTROL FOR NAT
This section serves as a general guidance for the development of laboratory- and procedure-specific QC and QA procedures for NAT. Aspects such as waste management, management of radioactive material, or working with hazardous material are not covered. NAT is a technology that offers extreme sensitivity with its ability to generate millions of amplicons from as little as a single nucleic acid template, resulting in a detectable signal. The advantages of this technology can be offset by the necessity of establishing complex assay protocols and the requirement to follow carefully very stringent QC/QA protocols. Deviation from these protocols can cause major problems, such as false positive results due to the contamination of templates by amplicons generated in previous assay runs. Similarly, failure to control inhibitors could lead to suboptimal amplification and possible false negative results. Given the myriad factors that can greatly influence the outcome of a NAT assay, all aspects concerning NAT need to be covered by appropriate and stringent QC/QA procedures. This requires careful facility design, workflow, and selection of equipment suitable to the purpose. Data recording, record keeping, and data interpretation are other aspects that should be covered by QC/QA. Thus, QA for NAT assays includes assay validation, establishment of acceptance criteria and specifications, and adherence to good manufacturing/laboratory practices. These aspects are also described in this section. In addition, reference should be made to other published guidelines such as the ICH Guideline Validation of Analytical Methods: Methodology (Q2B) and the NCCLS Guidelines.
Laboratory QC/QA
An NAT laboratory should be designed and operated in a manner that prevents contamination of reactions with products from previous amplifications (carry-over) as well as cross-contamination between samples. Historically, the application of PCR required strict separation of the various steps of the assay in order to prevent cross-contamination of PCR by amplicons. This was necessary because early procedures for analysis of PCR products involved the transfer of the product, which potentially could lead to contamination. Therefore, in an open system the best measure to prevent contamination has been the strict separation of working areas for individual process steps. This includes individual areas for template preparation, master mix setup, distribution of the master mix to individual reaction wells and addition of template, space for cycling the PCR assays and, optionally, a separate work space for PCR product analysis. These requirements are not necessary with closed systems. With both open and closed systems it is still necessary to take additional precautions. These safety measures include UV illumination of work spaces overnight to inactivate residual DNA by crosslinking. In case of contamination, laboratory benches and pipettes can be decontaminated by cleaning with a 10% solution of commercial bleach, which usually contains about 5% sodium hypochlorite, taking appropriate safety measures such as wearing gloves and eye protection. Afterwards, benches and pipettes should be rinsed with distilled water. A unidirectional workflow will reduce the opportunity for contamination to occur. Also, no materials, supplies, or equipment should be exchanged between designated working areas or rooms.
Equipment QC/QA
Other good laboratory practices that are related to the prevention of carry-over contamination include the use of suitable and clean equipment. Generally, disposable consumables (tubes, pipette tips, etc.) are highly preferable to reusable equipment. The use of disposable tips containing hydrophobic filters is another very effective measure to minimize cross-contamination. All samples, primer, probes, etc. must be labeled with relevant information such as identity of the content, date of use or preparation, expiration date, concentration, and storage information. Dedicated laboratory coats or disposable lab coats should be available in each room (or section) of the NAT laboratory. Appropriate gloves should be used during all processing steps to prevent sample contamination. The gloves should be changed frequently. Because heat sterilization does not completely destroy DNA, PCR products may lead to detectable contamination of, for example, glass surfaces. Following unique sterilization procedures for different materials such as waste and glass laboratory equipment is advisable.
Carry-Over Prevention with Uracil-N-Glycosylase
Contamination by PCR product carry-over can be mitigated by using the commercially available uracil-N-glycosylase (UNG) procedure. The procedure involves substituting 2¢-deoxyuridine 5¢-triphosphate (dUTP) for 2¢-deoxythymidine 5¢-triphosphate (dTTP) in the PCR setup and treating all PCR mixtures with UNG prior to PCR amplification, which can be easily incorporated as a first step into PCR cycling programs. Incorporating dUTP into the amplicon makes the PCR products biochemically distinct from the native DNA template. The enzyme UNG cleaves the deoxyuridine-containing PCR products by opening the deoxyribose ring at the C1 position. When the deoxyuridine-containing DNA is heated during the first thermal cycle, the amplicon DNA chain breaks at the position of the deoxyuridine at the alkaline pH of the PCR reaction mixture and thereby renders the carried-over PCR product nonamplifiable. Thus, any previously generated U-containing amplicon that might have contaminated another sample will become nonamplifiable. As a consequence, false positive results can be avoided. However, it should be noted that UNG has concentration limits above which it does not fully remove PCR carry-over products.
Validation of NAT Systems
Assay validation is achieved by
- ensuring the quality and consistency of assay components, including primers, probes, and enzymes; (including shelf life and contamination control) and
- establishing the performance characteristics of the NAT assay in terms of reproducibility, accuracy, ruggedness, robustness, specificity, precision, and analytical and clinical sensitivity.
The analytical sensitivity of an assay is defined as the minimum concentration of a reference reagent or standard detected by the test while the clinical sensitivity of a test is determined by testing clinical specimens and determining the 95% LOD. The clinical sensitivity of a test is not necessarily the same as the analytical sensitivity. The closer the reference or standard material is to the samples being tested the closer the correlation.
The principal steps of assay validation are
- sample preparation;
- consistent production of critical reagents;
- use of controls, calibrators, and quantitation standards;
- specimen and reagent stability;
- functionality of instruments and software;
- operator training; and
- laboratory surveillance for proficiency.
Following assay validation, further QA is necessary to monitor specifications and functional characteristics that have been established by the use of well-characterized reagents of known potency.
Quality Control of Reagents
DNA Templates—
The test specimens used are usually, but not limited to, whole blood, plasma, and serum. Specimen preparation is a key step in the NAT assay and has a major influence on the performance and variability of the assay. Specimen collection is the first step in sample preparation. QC/QA staff should carefully evaluate the effects on the integrity of DNA of collection tubes and temperatures during sample transport. To prevent cross-contamination during specimen collection, aseptic techniques should be used along with closed sampling systems in order to avoid specimen contamination. The use of appropriate sample handling techniques, temperature conditions, and anticoagulants or preservatives should help reduce the risk of contamination. Anticoagulants such as heparin or EDTA may interfere with the NAT assay.
Sample Extraction—
The buffers, reagents, and detergent or chaotropic agents used for nucleic acid extraction should be evaluated for inhibitory effects on the NAT assay. Extraction controls, including spiked materials, should be included to monitor the efficiency and reproducibility of the extraction method. Reproducibility of the sample preparation method should be determined under the specimen processing conditions, including sample handling, storage, and shipping conditions. DNA is generally stable, but personnel should take care to avoid storage at refrigerated temperatures for extended periods of time to avoid sample degradation. Repeated freeze–thaw cycles can sometimes cause DNA fragmentation. In the case that the target is RNA, it should be noted that RNA is very unstable and specimens should be frozen.
Primers—
Primers and probes should be qualified in terms of purity, identity, and functional potency. Purity can be assessed by use of HPLC or mass spectrometry; identity can be established by sequencing; and functionality can be established by the use of reference reagents. However, in many cases, these methods may not be available for in-house testing. In these cases, it may be sufficient to compare lot-to-lot variation of purity and functional potency using relevant methods available in-house coupled with the use of reference reagents.
DNA Polymerases—
The functionality of enzymes should be determined using reference materials. Enzyme preparations should be tested for other enzymatic activities; for example, exonucleases and DNA- and RNA-dependent polymerase activities and specifications should be established. Lot-to-lot comparison, as well as comparison with the manufacturer's CoA should also be done. Storage conditions recommended by the manufacturer should be strictly followed, and appropriate controls should be used to monitor the stability of enzymes.
Run Controls
The use of controls affords the operator assurance that the assay has performed within accepted specifications. In PCR testing, several steps in the testing process, as outlined above, should be monitored and verified. Multiple controls or controls that serve multiple purposes may be needed for a PCR assay. Controls should reflect the specific technology under development but should typically allow monitoring of ultracentrifugation, extraction, amplification, hybridization, quantitation, contamination, etc. Controls should be similar to the specimen type whenever feasible, although spiked controls may be acceptable.
A negative control is one that does not contain the target sequence or pathogen that is being tested. It should resemble as closely as possible the sample matrix under testing. Multiple negative controls should be examined, including nontarget sequences and nucleic acid-free controls to monitor for false positives resulting from contamination. Because of the high sensitivity of amplification assays, QC/QA personnel highly recommend that sponsors include control measures for the prevention of contamination events.
A positive control is one that contains the target sequence of interest. It should resemble as closely as possible the specimen matrix being tested and should contain an appropriate and defined amount of target sequences. (e.g., kit control).
Specifications for both positive and negative controls should be provided, as well as validation data supporting the proposed assay cut-off/reporting threshold value or the assay's limit of detection. The laboratory should define the source of the controls and calibrators and have a plan for their continued renewal. Controls can be infectious or non-infectious. In the latter case, validation of viral inactivation should be provided.
Reagent controls are often referred to as blanks and could include samples that have no target sequence, no enzyme, no primers, etc. These controls provide additional information about problems encountered in PCR assays.
An internal control is added to each specimen to ensure the overall validity of the individual test results. Internal controls are used to verify sample extraction, amplification, and detection.
External Quality Assessment and Proficiency Testing
Quality assessment of the laboratory is achieved by participation in periodic competency assessment and laboratory proficiency programs. The latter should include the testing of reference reagents and well-characterized panels to measure the technical proficiency of operators. Therefore, care should be taken to prevent cross-contamination, to monitor workflow, and to ensure careful specimen and test sample handling. Evaluation of operator proficiency should include participation in competency and quality assessment programs. Each operator in a particular laboratory should participate in such programs and should demonstrate comparable results.
Data Management
Complete and consistent documentation of all activities performed and all data generated is necessary. Such documentation does not only require the maintenance of records of the data generated through sample testing but also information about reagents and equipment calibration and maintenance. Moreover, any alteration in the assay procedure needs to be introduced through a planned change control process and documented in such a way that change can be assessed by an independent party.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Anita Y. Szajek, Ph.D.
Principal Scientific Liaison 1-301-816-8325 |
(GCBA2010) General Chapters - Biological Analysis |
USP35–NF30 Page 739
Pharmacopeial Forum: Volume No. 33(5) Page 1005