INTRODUCTION
Bovine serum is the liquid fraction of clotted blood, obtained from an ox (Bos taurus, among others), that has been depleted of cells, fibrin, and clotting factors. Since the advent of modern cell culture, manufacturers of biological products have used bovine serum extensively as a cell culture growth supplement. Its rich nutritional composition of proteins, growth factors, hormones, amino acids, vitamins, sugars, lipids, trace elements, and other components supports a broad range of cell culture applications in research and commercial manufacture of vaccines, natural source and recombinant biologics (hereafter biologics), engineered tissues, and other emerging cell-based therapeutic products intended for human or veterinary use. The predominant type of serum used in research applications is Fetal Bovine Serum (FBS). Calf serum (from newborn and older animals) is used much less frequently, but because of its lower cost it may be widely used in commercial manufacturing.
As is the case with other animal-derived products, bovine serum carries a potential risk of introducing extraneous agents into cell culture. Serum manufacturers and regulators must adopt rigorous sourcing and testing procedures and strict processing and production guidelines to ensure the quality of bovine serum.
The objective of increasing the quality and safety of biologics produced with bovine serum, coupled with attempts to mitigate regulatory burden, have caused developers to investigate alternatives to serum supplementation, resulting in application-specific serum-free medium formulations. Although it is recognized that bovine serum should be avoided when there is an option to use serum-free medium, there are cases where this is technically impossible or impractical.
This chapter describes issues related to sourcing, production, and characterization of bovine serum to ensure its safe use. A list of relevant regulatory and guidance documents is presented in Appendix 1. Serum manufacturers and serum end users (manufacturers of biological products) should consider and apply as needed the controls and procedures outlined in this chapter to ensure the safe use of bovine serum components in research and pharmaceutical manufacturing.
Types of Bovine Serum
- FBS is obtained from the fetuses of healthy, prepartum bovine dams that had been deemed fit for human consumption through ante- and postmortem inspection by licensed veterinarians. It is collected in government-inspected and -registered slaughterhouses.
- Newborn calf serum (also known as newborn bovine serum) is obtained in government-inspected and -registered slaughterhouses from animals aged less than 20 days.
- Calf serum is obtained in government-inspected and -registered slaughterhouses from animals aged between 20 days and 12 months.
- Donor bovine serum (also known as donor calf serum) is obtained by the repeated bleeding of donor animals from controlled government-inspected and -registered donor herds. The animals are 12–36 months old.
- Adult bovine serum is obtained in government inspected and -registered slaughterhouses from cattle older than 12 months that are declared fit for human consumption.
BOVINE SERUM: HISTORY AND TYPES OF USE
History of Bovine Serum Use
Animal serum and other complex biological materials have been employed in the cultivation of mammalian cells for approximately 100 years. Several factors led to the wide adoption of bovine serum as a standard tissue culture supplement. In comparison to serum from other animal species (horse, goat), bovine serum is easily sourced, and thereby more affordable. Many investigators choose to use fetal serum in their experimental systems because of concerns associated with antibodies present in newborn and adult serum that could cross-react with cells in culture and cause cell lysis through complement-mediated pathways. To eliminate that concern, heat was introduced to inactivate complement that was potentially present in the serum. Studies of FBS undertaken in the 1950s on the cultivation of low-density human cells to elucidate mechanisms of cell growth found that (1) the albumin component may serve as a carrier of essential small molecules; (2) fetuin, a glycoprotein present at high levels in the alpha globulin fraction, facilitates cell attachment and stretching; and (3) fetuin markedly inhibits trypsin, and this antiproteolytic activity may play a role in the ability of fetuin to stimulate cell growth.
In the 1960s and 1970s, serum supplementation of tissue culture media became the norm, thus facilitating biomedical research as well as the first large-scale vaccine manufacturing processes. Serum supplementation reduced the requirement for optimizing medium formulations for different cell types. FBS was shown to provide a variety of polypeptide growth factors. Albumin promoted cell growth presumably because of its abilities to function as a carrier protein for small molecules or lipids, to bind metal ions, to serve as a pH buffer, and to protect cells against shear. Similar functions were found for other serum components such as transferrin, hormones, and other serum-derived attachment factors such as fibronectin, vitronectin, and laminin.
Uses of Bovine Serum
Serum is a complex mixture of macromolecules that is required for cell growth and virus production, and its use as a raw material presents a number of challenges. These include its batch-to-batch composition and the risk of contamination by adventitious agents. The development of serum-free media has replaced serum in some new biotechnology manufacturing applications, but many cell lines used in manufacturing have not been adapted to these serum-free media. Regulatory constraints and scientific challenges generally make it impractical to alter existing manufacturing processes in which serum is used as a raw material.
FBS sometimes is required in cell and tissue bioprocessing, which often involves the cultivation of cells from tissue explants and biopsies. Some bioprocesses may also require the maintenance of specific cellular characteristics during cultivation. FBS often appears to facilitate such procedures and may affect the biological behavior of fastidious cell types. FBS has been shown to affect the transcription of developmentally important genes, apoptosis, and apoptosis-related gene expression, and to provide neuroprotective and antioxidative factors, all of which may be beneficial to the survival and development of cells in culture. Therefore, FBS will continue to play an important role as a cell culture supplement for production of cell- and tissue-based therapies.
In most viral vaccine manufacturing processes the media used for cell culture expansion and virus infection/production are supplemented with different types of serum at different concentrations. In these processes, bovine serum helps generate a mass of cells in an optimal physiological state for efficient viral replication.
BOVINE SERUM HARVESTING AND PRODUCTION
Blood Collection
For all types of bovine sera, blood should be collected in government-inspected and -registered premises (slaughterhouses, abattoirs, and donor farms). Blood should be collected by trained operators following the written procedures approved by the serum manufacturer and using either single-use disposable collection devices or reusable collection equipment for which cleaning procedures have been validated.
Donor Bovine Serum
For each lot of serum from donor animals, serum manufacturers should ensure traceability to the donor herd of origin via production records and animal health and origin certificates. Donor animals are subjected to regular veterinary inspections and are bled multiple times following established procedures. Animals introduced into the herd should be traceable by source, breeding, and rearing history. Collectors should introduce new animals into the herd following specified and approved procedures that include prepurchase animal inspection and testing, proper transportation, a quarantine period, veterinary examination and testing during the quarantine period, and animal release criteria from quarantine to serum production. The collectors should not vaccinate donor animals for bovine viral diarrhea (BVD). Collectors should test animals for any agent and antibody from which the herd is claimed to be free.
Newborn Calf Serum, Calf Serum, and Adult Bovine Serum
Certificates of animal health and origin and/or serum production records should ensure that serum manufacturers can trace bovine serum derived from slaughtered animals back to the abattoir. Serum manufacturers should require abattoirs to maintain documentation of the origin of animals for slaughter. Blood should be collected from animals that have been slaughtered, for human consumption, in abattoirs inspected by the competent authority of the country of origin. Inspectors should routinely inspect animals both antemortem and postmortem to check for the clinical appearance of infections and parasitic diseases and other animal health-related problems or conditions. The animals must be free of clinical evidence of infectious diseases at the time of slaughter. Blood collection procedures must be in place to prevent cross-contamination with other tissues and body fluids and the surrounding environment. The standard procedure of slaughter consists of an approved method of animal stunning followed by exsanguination.
Fetal Bovine Serum
FBS product specifications and test procedures are presented in the proposed general chapter Fetal Bovine Serum—Quality Attributes and Functionality Tests  90
90 . Serum manufacturers should collect fetal bovine blood from bovine fetuses whose dams have been slaughtered. The dams must have been deemed fit for human consumption and must have been slaughtered in abattoirs that were inspected by the competent authority of the country of origin. Inspectors should examine all animals both antemortem and postmortem to check for the clinical appearance of infections and parasitic diseases and other animal health-related problems or conditions. The animals must be free of clinical evidence of infectious diseases at the time of slaughter. The uterus is removed and transported to a dedicated space for fetal bovine blood harvest, where blood collection personnel evaluate the fetus for signs of fetal death, including bloating, skin discoloration, odor, deformation, and hair sloughing. Collectors also should check the amniotic fluid for color, quantity, and clarity. Serum manufacturers should collect blood from acceptable fetuses by cardiac puncture into a closed collection system under conditions designed to minimize microbial contamination. Manufacturers should have in place procedures that will prevent cross-contamination with other fetal tissues and bodily fluids and the surrounding environment.
. Serum manufacturers should collect fetal bovine blood from bovine fetuses whose dams have been slaughtered. The dams must have been deemed fit for human consumption and must have been slaughtered in abattoirs that were inspected by the competent authority of the country of origin. Inspectors should examine all animals both antemortem and postmortem to check for the clinical appearance of infections and parasitic diseases and other animal health-related problems or conditions. The animals must be free of clinical evidence of infectious diseases at the time of slaughter. The uterus is removed and transported to a dedicated space for fetal bovine blood harvest, where blood collection personnel evaluate the fetus for signs of fetal death, including bloating, skin discoloration, odor, deformation, and hair sloughing. Collectors also should check the amniotic fluid for color, quantity, and clarity. Serum manufacturers should collect blood from acceptable fetuses by cardiac puncture into a closed collection system under conditions designed to minimize microbial contamination. Manufacturers should have in place procedures that will prevent cross-contamination with other fetal tissues and bodily fluids and the surrounding environment.
Serum Harvesting and Processing
Trained personnel should perform serum separation (harvesting) and further processing activities following written and approved procedures. Serum is first separated and pooled, followed by filtration and filling into clean and disinfected containers. If the serum is subjected to one or more virus inactivation treatments in the production process, serum manufacturers should validate the virus inactivation processes against a range of relevant viruses. It is recommended that bovine viral diarrhea virus (BVDV) be included in any virus validation study because it is ubiquitous.
Serum Separation and Harvesting
Bovine blood should be processed and serum separated (harvested) in such a way as to minimize bacterial and mycoplasmal contamination, which in turn minimizes endotoxin levels in serum product. Gentle, quick blood processing helps to minimize hemolysis, further enhancing the quality of the serum product. After collection, blood is first allowed to clot for a specified period of time and under controlled conditions, then centrifuged in a refrigerated centrifuge. Serum is then removed from the clot, typically by centrifugation; pooled and mixed in a pooling vessel; transferred to labeled containers; and frozen, unless it is filter-sterilized immediately. Serum manufacturers should describe each process step and carry out serum processing activities, including sample collection and in-process quality control testing, following the manufacturer's approved procedures.
Pooling Before Filtration
Because limited amounts of blood can be collected from individual animals, serum manufacturers pool the raw serum from many animals in order to create commercial-sized lots. Serum is pooled, after raw serum thawing and before filtration, in a pooling vessel and mixed at a controlled mixing rate and temperature. Pools or lots of donor bovine serum may consist of many separate collections from individual members of the herd. Lots of FBS may consist of pooled serum from thousands of animals. Serum manufacturers should describe each prefiltration pooling process step and should carry out serum thawing, prefiltration pooling, and mixing activities following the manufacturer's approved procedures.
Filtration
Pooled serum is mixed and aseptically passed through filters of pore size 0.2 µm or smaller, depending on the intended application. Filtration processes should be validated. Triple filtration using filters of pore size 0.1 µm has been shown to result in a high degree of mycoplasma removal. Although filtration may remove some large viruses and viral aggregates from the serum, generally viruses cannot be completely eliminated in this manner. Furthermore, the filters are not known to eliminate the causative agent of bovine spongiform encephalopathy (BSE). Following filtration, serum manufacturers fill filtered serum into sterile containers by aseptic processing in a suitably controlled environment. Serum manufacturers should describe each filtration process step and should perform serum filtration, filling, and sample collection activities following the manufacturer's approved procedures.
Irradiation
Serum treatment by gamma irradiation is very common and one of the most effective methods of virus inactivation. The most frequently used minimum dose is 25 kilograys (kGy). Some countries specify higher dose requirements  (>30 kGy) for imported serum. Gamma irradiation may inactivate viruses, mycoplasma, and bacteria, but serum end users should ensure that the gamma irradiation process does not negatively affect their specific applications. Irradiation may have adverse effects on serum quality, and these adverse effects tend to increase with higher doses.
(>30 kGy) for imported serum. Gamma irradiation may inactivate viruses, mycoplasma, and bacteria, but serum end users should ensure that the gamma irradiation process does not negatively affect their specific applications. Irradiation may have adverse effects on serum quality, and these adverse effects tend to increase with higher doses.
Validation of gamma irradiation has two aspects: (1) dose delivery in a defined load configuration and (2) inactivation capacity. Critical irradiation process parameters include product (serum) temperature, packaging size and configuration, dosimeter distribution, and defined minimum/maximum dose received. Dosimeters should be used to monitor the established high-dose and low-dose positions in each irradiation run. If the serum manufacturer makes inactivation claims, these should be supported by the manufacturer’s own well-designed viral inactivation studies.
Ultraviolet (UV) Treatment
Serum manufacturers may use UV treatment to inactivate viruses, mycoplasma, and bacteria, but manufacturers must validate the process to demonstrate its efficacy. In addition, manufacturers must be aware that UV treatment may have an adverse effect on serum quality and accordingly should consider the effects of UV treatment for each application, as should serum end users.
Heat Inactivation
Heat inactivation involves elevating the temperature of the serum to >56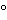 for at least 30 minutes to inactivate complement. Heat inactivation may also inactivate viruses, mycoplasma, and bacteria; but it may have an adverse affect on serum quality, and manufacturers must validate the procedure’s suitability for specific applications. Heat inactivation provides significantly less assurance of virus inactivation than does irradiation.
for at least 30 minutes to inactivate complement. Heat inactivation may also inactivate viruses, mycoplasma, and bacteria; but it may have an adverse affect on serum quality, and manufacturers must validate the procedure’s suitability for specific applications. Heat inactivation provides significantly less assurance of virus inactivation than does irradiation.
Viral Clearance Studies
Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin 1050 and other regulatory documents give guidance about conducting viral clearance studies that help validate removal/inactivation processes. Serum manufacturers should also perform formal spiking studies with relevant and representative (model) viruses, and should test and compare inactivated spiked serum samples, negative controls, and positive controls.
Charcoal Stripping
Some serum manufacturers use charcoal/dextran treatment to reduce the levels of hormones in serum.
Dialysis
Some manufacturers use dialysis or diafiltration to remove low molecular weight components from serum.
Cleaning and Sterility of Equipment
Stainless steel systems and tubing used in the manufacture of bovine serum must be cleaned and sterilized to prevent cross-contamination and growth of adventitious agents. Serum manufacturers must validate their cleaning processes for removing and inactivating agents of concern. Thereafter, manufacturers should implement process controls that routinely verify cleaning cycles. Steam sterilization-in-place is a common and effective sterilization technique. Serum manufacturers that use this technology must validate steam cycles to demonstrate their uniformity and ability to destroy heat-resistant bacterial spores. Alternatively, manufacturers can use sterile disposable systems that do not require cleaning validation.
Quality Control
Traceability
Abattoir Collection
Materials collected in the U.S. should originate from U.S. Department of Agriculture (USDA)-registered facilities. Serum manufacturers should maintain documentation that traces a given serum sub-lot to the abattoir where it was collected. Slaughterhouses maintain records of animal source. General industry practice is to keep this information as part of the Device Master Record. General record-keeping requirements at USDA-licensed abattoir facilities are outlined in 9 Code of Federal Regulations (CFR) 320.
Materials collected from countries approved by the USDA for importation of bovine products into the U.S. should meet the requirements of the competent authority of the country of origin. In addition, serum manufacturers should keep USDA-required safety testing records of imported materials (if applicable) as part of their Device History Record.
Serum manufacturers should consult 9 CFR 309 and 9 CFR 310 about requirements for inspection of animals for various diseases pre- and post-slaughter. These requirements are recommended for materials collected outside the U.S.
Donor Herd Collection
Serum manufacturers should maintain traceability to the donor animal farm where blood was collected from donor animals. In most cases, manufacturers individually identify farm animals and keep records for bleed and processing dates, making it possible to trace blood collection to an individual animal. A licensed veterinarian or a designee under the guidance of a veterinarian should inspect animals regularly and should certify that the animals are free of disease and fit for human consumption, consistent with 9 CFR 309.
Product Storage and Stability
Serum should be stored in the frozen state at 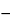 10 or below. Serum is frozen as quickly as possible to preserve product quality and is stored under controlled storage conditions. Serum manufacturers should establish serum product stability in support of a proposed expiration date. Typical expiration dating for bovine serum is 5 years from the date of filtration and filling. Use of any type of bovine serum beyond the stated expiration date depends on the application, and the serum user must establish the product’s continued suitability for use.
10 or below. Serum is frozen as quickly as possible to preserve product quality and is stored under controlled storage conditions. Serum manufacturers should establish serum product stability in support of a proposed expiration date. Typical expiration dating for bovine serum is 5 years from the date of filtration and filling. Use of any type of bovine serum beyond the stated expiration date depends on the application, and the serum user must establish the product’s continued suitability for use.
Labeling
Finished product labels must contain the following information: product description, lot number, storage conditions, name and address of manufacturer, and a statement indicating the intended use. Materials intended for research purposes are exempt from labeling regulations (21 CFR 801). Typically, serum manufacturers supply a lot-specific Certificate of Analysis (COA) that is classified as part of the product’s labeling. See COA requirements in the following section.
Certification/Documentation
Certificate of Analysis
The COA should provide information about a specific lot of serum, including tests performed and test results (according to the serum manufacturer’s specifications for release), as well as critical labeling identifiers such as lot number, catalog number, description of type of bovine serum, country of origin, and either or both dates of manufacture and expiration. This document is distinct from the certificate of health issued by the competent authority of the country of origin.
Certificate of Origin and Certification of Animal State of Health
The Certificate of Origin establishes the country in which the bovine blood was collected and veterinary certification of the health of the animals pre- and postcollection (9 CFR 327.4).
Import/Export Documents
Import/export documents contain formal certification of animal disease status of the country of origin and negotiated/agreed certification statements. These vary from country to country. Each country defines import/export requirements in order to control introduction of exotic animal diseases and their economic impact as well as product safety assessments (risk vs. research, diagnostic, and/or therapeutic benefits).
Production Reports
Production reports typically are batch records that document the raw materials in identifiable and traceable ways, production methods (centrifugation or filtration) used in manufacturing, equipment and facility cleaning, quality control testing, and personnel performing required activities. Raw material with Certificates of Origin or serum production records facilitates traceability to the source of the blood that was used to create the serum. When serum is used as a raw material for further manufacturing, process documentation also helps demonstrate controlled manufacture of the bovine serum.
BSE RISK ASSESSMENT
Despite the low risk potential of transmissible spongiform encephalopathies (TSEs) in bovine serum, various U.S. and international regulatory agencies have developed guidance to help manage and further reduce the potential risks of transmission. In the absence of appropriate test methods of detecting the infectious agent in fluids such as blood, the consensus recommendation from various regulatory agencies is to adopt good risk assessment strategies. This section of the chapter provides some background information on the disease and current methods of detection; it also highlights risk assessment and risk reduction strategies to potentially prevent transmission of the disease through the use of serum in the manufacturing of medicinal products.
Description of the Disease
TSEs are transmissible animal and human diseases that are characterized by degeneration of the brain, associated with severe neurological signs and symptoms. Since the outbreaks of TSE in cattle, termed BSE, which were transmitted to other species, public health officials have been concerned about the risk of TSE infection, including the possibility of TSE transmission by the use of therapeutic products manufactured using bovine serum. In cattle infected with BSE, lower titers have been found in the cerebrospinal fluid, lung, lymph tissue, spleen, kidney, liver, and ileum. Studies have shown that transfusion of blood from sheep infected with either BSE or scrapie but without evident disease can infect naive sheep. Although the risk of cross-contamination is always present, to date no studies have shown that blood can transmit disease from cattle with BSE. Embryos from BSE-affected cattle have not transmitted diseases to mice. Calves born of dams that received embryos from BSE-affected cattle have survived for up to 7 years, and examination of the brains of both the unaffected dams and their offspring revealed no spongiform encephalopathy.
Detection Strategies
No currently available procedures have been validated as being sufficiently sensitive for routine antemortem screening of asymptomatic animals, although analytical methods are under development for detection and quantitation from low-infectivity materials such as blood. The classic diagnostic test for TSEs is postmortem histological examination of brain tissue to confirm characteristic vacuolar degeneration. Other testing options include immunohistochemical tests that can confirm the presence of PrPSc, the abnormal disease-specific conformation of prion-related protein (PrP), in the vacuolated regions of the brain; and immunochemical tests such as Western blots and enzyme-linked immunosorbent assays that can detect PrPSc in tissues with high or moderately high titers. These tests typically take less time to perform than histological examination (6–8 hours vs. weeks, respectively) and can be partially or fully automated. Although most of these are postmortem tests, studies have demonstrated the feasibility of antemortem testing of lymphoid tissue samples from the tonsils or from the third eyelid of infected animals. Immunochemical tests require extensive sample collection and preparation and can be cost prohibitive for routine testing and monitoring the disease state of large herds. Diagnostic strategies must consider the sensitivity of testing in certain tissues as well as the test’s ability to detect infectivity in animals before the development of clinical signs of disease. Negative results do not ensure the absence of infectivity. Detection of infectivity is possible if suspect tissue is inoculated into experimental animals intracranially where the causative agent can amplify. This approach for detection of low infectivity can take months to years to yield a positive result.
Risk Assessment and Risk Reduction Strategies
Serum manufacturers should employ risk reduction strategies to eliminate the danger of cross-contamination of fetal blood with other tissues, including appropriate sourcing of animal-derived articles and using practices that have been shown to eliminate or minimize the risk of transmitting TSE, via either foods or health care products. Serum end users should perform a risk assessment of their sourcing strategy that takes into account the amount of bovine serum used in their application and should conduct supplier audits to ensure traceability of sourcing, handling, and appropriate quality control systems.
Source and Age of Animals
Serum manufacturers should monitor the traceability of each lot of serum to ensure the qualification of bovine serum, as described previously in the two sections Serum Harvesting and Processing and Quality Control. In addition to traceability, careful selection of source materials is the most important criterion for the safety of medicinal products. Certification of the origin must be available from the supplier, and manufacturers should keep this information on file. The U.S. Food and Drug Administration (FDA) recommendations prohibit the use in FDA-regulated products (except gelatin) of any bovine-derived materials that originate from countries that report indigenous cases of BSE. The current proposed rule qualifies FBS as an unlikely source of BSE infectious material, because current evidence suggests that cow-to-calf transmission of BSE is unlikely. The proposed rule also states that prohibited cattle materials do not include materials sourced from fetal calves of cows that were inspected and passed, as long as the materials were obtained by procedures that can prevent contamination with specified risk materials. For veterinary biologics, current regulations enforced by the USDA's Center for Veterinary Biologics (CVB) indicate that ingredients of animal origin should be sourced from countries with no or low BSE risk, as defined by the U.S. National Center for Import and Export and 9 CFR 94.18.
The most satisfactory sources of materials are from countries with the following:
- No reported cases of indigenous BSE
- Compulsory notification of positive tests
- Compulsory clinical and laboratory verification of suspected cases
- Prohibition of the use in ruminant feed of meat and bone meal containing any ruminant protein
- No importation of cattle from countries where a high incidence of BSE has occurred
- No importation of progeny of affected females
BSE infectivity may increase with animal age. Although bovine serum is considered a low-risk material for TSE transmission, some end users consider it prudent to source serum from dams below a set maximum age. If manufacturers cannot determine the date of the dam’s birth, they should consider both the implementation date of the feed ban in the country of origin and the incubation period of BSE in order to determine the safety of the source. A ruminant feed ban was imposed in the United Kingdom in July of 1988. These considerations are lot specific, so audits of the raw material supplier should include a review of records.
Production Process
End user manufacturing systems should be in place for monitoring the production process and for batch delineation (definition of batch, separation of batches, and cleaning between batches). Of primary importance is control of the potential for cross-contamination with possible infectious material. Because of the documented resistance of TSE agents to most inactivation procedures, controlled sourcing is the most important criterion in achieving acceptable product safety.
Whenever possible, manufacturers should identify steps that theoretically or demonstratively remove or inactivate agents during the manufacture of the material. Manufacturers should continue their investigations into removal and inactivation methods to identify steps/processes that will help ensure the removal or inactivation of TSE agents. Manufacturers should design production processes using available methods that have the greatest likelihood of inactivating or removing TSE agents. For example, prolonged exposure of tissues to high moist heat and high pH inactivates the BSE agent. Such treatments, however, are inappropriate for the extraction of many other types of bovine-derived articles, such as serum, because these treatments lead to the destruction of the material. Conventional chemical and biochemical extraction and isolation procedures may be sufficient to remove the infectious agent. Similar techniques may be effective for other bovine-derived articles. Further research will help to develop an understanding of the most appropriate methodology for validation studies. Issues to consider during validation of a process for removal of TSE agents include the following:
- The nature of the spiked material and its relevance to the natural situation
- Design of the study (including scale-down approaches)
- Method of detecting the agent (in vitro or in vivo assay) after spiking and after the treatment
- Characterization and standardization of reference materials for spiking
-
Data treatment and analysis (see Design and Analysis of Biological Assays 111)
Because no studies have successfully validated analytical methods for the detection of small amounts of the TSE agent, TSE clearance validation studies typically employ the intracranial injection of in-process material into rodents for amplification and detection of potential residual infectivity.
TESTING AND CONTROL OF ADVENTITIOUS AGENTS
Introduction
Rigorous testing procedures, strict processing and production guidelines, and appropriate risk assessments help ensure the safety of the different types of bovine serum. This section discusses specific tests that can detect and control adventitious agents.
Adventitious Agents Testing
The adventitious agents testing required for the evaluation of master seeds, master cells, and bulk and final products is described in 9 CFR 113.53 and by directives from the European Agency for the Evaluation of Medicinal Products (EMEA) (EMEA/CVMP/743/00 and EMEA/CPMP/BWP/1793/02). The testing methods outlined in these documents can detect a wide range of bovine microbial agents in serum products. These testing methods meet the requirements for most of the world’s regulatory agencies. Serum manufacturers should test a representative sample of each batch of serum to determine the presence of adventitious agents. Testing is performed after filtration but before any further processing that is intended to inactivate or remove viruses.
Filtration with 100-nm (0.1-µm) pore size filters is an accepted method for removing mycoplasmas and gamma irradiation (> 25 kGy while frozen), and chemical treatments (e.g., with betapropiolactone) are accepted methods of inactivating viruses and mycoplasmas; serum manufacturers routinely use these tools in both production and testing facilities. These treatments do not remove antibodies that may interfere with some applications. Additionally, the treatments do not ensure complete viral removal or inactivation, but can significantly reduce the risk of viral activity. The testing series to screen bovine serum for the absence of adventitious agents typically includes the following:
- Bacterial and fungal sterility testing as described in 9 CFR 113.26
- Mycoplasma testing as described in 9 CFR 113.28
- Viral testing as described in 9 CFR 113.53
The procedures described in Sterility Tests 71 confirm the absence of bacterial and fungal infection. For viruses, only cultivation using suitable substrate cells can indicate viral infectivity and replication. Those who use serum for research or production should test the serum for the absence of adventitious agents in a manner that is consistent with the product's intended application, bearing in mind that testing indicates only presence or absence of adventitious agents within the limits of the test procedures used.
Mycoplasma Testing
Mycoplasma contamination in tissue culture can arise from many animal origin sources, including serum, but more commonly it results from cross-contamination of infected cultures. Mycoplasmas are particularly insidious contaminants in cell culture because they
- cannot be visualized by light microscopy even at high density (>107 colony-forming units/mL);
- cause no observable change in turbidity or pH of the culture fluid;
- cannot routinely be removed by single sterilizing filters, although removal can be obtained through a triple series of 0.1-µm filters;
- are unaffected by traditional antibiotics used in cell culture; and
- exert an extremely wide variety of adverse effects in tissue culture.
Classical mycoplasma detection is described in Mycoplasma Tests 63.
In addition to these methods, more recent detection procedures include luminescent and polymerase chain reaction (PCR) assay procedures. Nucleic Acid-Based Techniques—Amplification 1127 describes the general principles of PCR assays. The sensitive 20-minute luminescent assay measures a specific enzyme activity of mollicutes that converts adenosine diphosphate to adenosine triphosphate via a luciferase/luciferin reaction. Results are unequivocal and semiquantitative. PCR methods are quick and sensitive and display with good reliability, but occasional false positive results are a source of concern with commercial testing service labs. PCR may detect mycoplasmal DNA fragments that are non-infectious.
Viral Testing
The virus testing procedures for serum products are outlined in 9 CFR 113.52 and 9 CFR 113.53. In addition, there are other documents that may include equivalent or relevant testing such as EMEA/CVMP/743/00-Rev.2 from the Committee for Veterinary Medicinal Products (CVMP) Revised Guideline on Requirements and Controls Applied to Bovine Serum Used in the Production of Immunological Veterinary Medicinal Products and EMEA/CPMP/BWP/1793/02 from the Committee for Proprietary Medicinal Products (CPMP) Note for Guidance on the Use of Bovine Serum in the Manufacture of Human Biological Medicinal Products. Serum manufacturers should perform virus testing in compliance with this regulation, using at least two different and sensitive detector cell lines, one of which should be of bovine origin. The tests include cultivation of detector cells in cell culture media supplemented with 15% test serum for at least 21 days. Cells are subcultured at least twice during this period, usually 7 and 14 days post inoculation. At the conclusion of the last subculture (after a total of at least 21 days of incubation), cells are examined for general signs of virus amplification. The following end points are used for general virus detection: microscopic cell examination for cytopathogenic agents such as infectious bovine rhinotracheitis virus, cell staining and microscopic examination for inclusion bodies, and hemadsorption test to detect hemadsorbing agents such as PI-3. In addition to this series of testing and at the conclusion of the last subculture (after a total of at least 21 days of incubation), cells are stained with specific fluorescent antibodies against the following specific viral agents:
- BVDV
- Bovine parvovirus
- Bovine adenovirus
- Bluetongue virus
- Bovine respiratory syncytial virus
- Reovirus
- Rabies virus
In addition to the viruses listed above, other viruses can be causative agents of disease and may require testing in various bovine serum applications. The serum end user is responsible for determining whether full 9 CFR testing is sufficient, and if other specific viral agents should be tested for. Examples of specific viruses not covered by the current virus testing guide may include akabane, bovine herpesvirus 1 (BHV-1), Parainfluenza-3 virus (PI-3), bovine leukemia, bovine rotavirus, bovine circovirus, bovine polyomavirus, coronavirus, torovirus, bovine enterovirus, bovine astrovirus, foot-and-mouth disease virus (FMDV), and rinderpest. Appendix 2 provides a general description of some of these viruses as well as the ones for which testing is required. A serum end user’s thorough risk analysis should determine the scope of testing and serum treatment options.
Risk Assessment and Detection Strategies
Serum manufacturers and serum end users should carry out a comprehensive, science-based risk assessment (e.g. Failure Modes and Effects Analysis) in order to better understand the safety profile of the serum product. The following risk assessment elements can be taken into consideration, but other elements can be included as appropriate: country of origin, region of the country, animal disease status of the country/region of origin, animal age, blood collection process, animal stunning method and exsanguination method, serum manufacturing process, type of production quality system, production in-process controls, final product testing, virus inactivation, equipment segregation, equipment cleaning procedure, personnel training, serum use/application, pharmaceutical product type, and intended use.
The species barrier provides a degree of protection against infection by some animal etiologic agents. This barrier is not an alternative to proactively ensuring that pharmaceutical products are manufactured only from raw materials of animal origin that have undetectable levels of adventitious agents. Inoculation of viable organisms into a nonhost species carries a risk that the organisms could cross the species barrier. An appropriate test regimen of serum material should therefore include examination for potential contaminants associated with the species of origin and the species of intent. Serum treatments to inactivate viral agents are a factor in establishing the appropriate test regimen for a particular material. Lowest risk of contamination is associated with biological materials that are terminally sterilized.
Zero risk is neither possible nor reasonable. The serum manufacturers should fully describe specific testing regimens in the product specifications, and these will vary depending on the type and source of the serum. Therefore, the guidelines for screening described in this chapter are examples only, and screening for all viruses listed may not be required for a particular material. Some manufacturers may perform certain tests on the finished product or on in-process materials rather than on individual component(s). Manufacturers must also evaluate the dilution effect in relation to the limit of detection of the test procedure. Interference with growth or neutralization of viral activity by serum may be an indication of a specific antibody or certain nonspecific factors in serum masking the viral agent. It is recommended that serum manufacturers consider this possibility when determining an adequate level of treatment in their viral inactivation studies or in virus testing applications.
Serum manufacturers should confirm that the species of origin is bovine to ensure that no other nonbovine agents may be present. Manufacturers should perform extraneous virus testing in appropriate cell cultures (see Virology Test Methods 1237 for appropriate cell line choices dependent on assay and targeted agent). If necessary, seroconversion studies should be conducted in susceptible animal species using a host species immune antibody response as the method of detection. Studies should use this procedure following an inactivation step to detect whether the virus was present before the virus inactivation process.
Serum manufacturing processes should be conducted in a consistent manner, following the established manufacturing procedures, with adequate quality systems built into the production process. Furthermore, equipment segregation (by species of origin), equipment and facility cleaning procedures, and personnel training are important elements in the risk assessment of the process.
Safety Considerations
End users of donor bovine serum may require serum that does not have detectable antibodies against BVDV or other specific agents so that the users can propagate cell cultures used in vaccine production, diagnostic testing, and test kit preparation, especially for the maintenance of master seed and master cell stocks. More than 40 cell types are available for the production of veterinary biologicals, but fewer than 10 media types are available for their propagation. Some researchers have proposed serum-free media as an alternative in propagating certain cells and viruses; but this means adapting culture procedures, which may alter the cells and change production results. If new or different sera are imported into the U.S., serum end users will require confirmation of source, species, and documentation of the origin of the sera in countries that are free of FMD and rinderpest.
CHARACTERIZATION OF BOVINE SERUM
Introduction
In the absence of end product-specific requirements, each lot of FBS should be tested to confirm that the serum meets the requirements of the proposed general chapter Fetal Bovine Serum—Quality Attributes and Functionality Tests 90. For all other types of bovine sera, this section describes several key procedures for characterization. These procedures are not mandatory but are guidelines that manufacturers may consider for their individual applications. The table in the Hemoglobin section shows samples of specifications for the different types of bovine sera.
Species Identification
Both inter- and intraspecies identification assays should be performed on bovine sera to confirm species identity and the integrity of the serum products, and to ensure that nonbovine agents are not present. The most commonly used assay for the identification of bovine species identity is based on the electrophoretic profile of specific serum proteins. With electrophoresis, the serum proteins usually separate into as many as six fractions: albumin, alpha 1, alpha 2, beta 1, beta 2, and gamma globulins.
Other procedures used for bovine speciation include radial immunodiffusion (RID) and the double diffusion Ouchterlony method. These procedures allow either qualitative or quantitative measurements of the immunoglobulin G levels in serum. The RID method is based on the diffusion of an antigen from a circular well into a homogeneous gel that contains specific antiserum for each particular antigen. A circle of precipitated antigen and antibody forms and continues to grow until it reaches equilibrium. The diameters of the rings are a function of antigen concentration. The Ouchterlony method is a double gel diffusion test wherein antigen and antibody diffuse toward each other in a semisolid medium to a point in the medium where optimum concentration of each is reached, forming a precipitate. The Ouchterlony plates contain cylindrical wells—a central 8-mm diameter antigen well, surrounded by six 3-mm antisera wells—which make possible the simultaneous monitoring of multiple antigen–antibody systems and the identification of particular antigens in a preparation. The proposed general chapter Fetal Bovine Serum—Quality Attributes and Functionality Tests 90 describes the accepted procedure.
Hemoglobin
Hemoglobin is a multi-subunit protein that forms an unstable reversible bond with oxygen in the red blood cells. The oxygen-loaded form is called oxyhemoglobin and is bright red. The oxygen-unloaded form is called deoxyhemoglobin and is purple-blue. Oxyhemoglobin is the predominant form in red blood cells.
Low hemoglobin content in sera is widely accepted as a good general indication of rapid and careful processing of blood that will be used for serum. Red blood cells are fragile and rupture easily, releasing hemoglobin into the serum. Rough handling of the harvested blood, poor temperature control, or delayed processing elevates hemoglobin content in serum. Acceptable levels of hemoglobin may vary with intended application. The hemoglobin levels are measured using spectrophotometric procedures (see Spectrophotometry and Light-Scattering 851) as described in the proposed general chapter Fetal Bovine Serum—Quality Attributes and Functionality Tests 90.
| FBS | Newborn Calf Serum |
Calf Serum | Donor Bovine Serum |
Adult Bovine Serum |
|
|---|---|---|---|---|---|
| Sterility test | No growth detected |
No growth detected |
No growth detected |
No growth detected |
No growth detected |
| Mycoplasma | Not detected | Not detected | Not detected | Not detected | Not detected |
| Virus testing | Not detected | Not detected | Not detected | Not detected | Not detected |
| Hemoglobin (mg/dL) | <30 | <30 | <30 | <30 | <30 |
| Total protein (g/dL) | 3.0–4.5 | 3.5–6.0 | 5.0–8.0 | 5.0–8.0 | 6.0–10.0 |
| pH | 7.00–8.00 | 7.00–8.00 | 7.00–8.00 | 7.00–8.00 | 7.00–8.00 |
| Osmolality (mOsmol/Kg) | 280–360 | 240–340 | 240–340 | 240–340 | 240–340 |
Chemical Profile
The testing of components such as cholesterol, alpha globulin, beta globulin, gamma globulin, albumin, creatinine, bilirubin, glucose, alanine aminotransferase, aspartate aminotransferase, phosphorus, potassium, calcium, and sodium usually is not considered a criterion for bovine serum lot release. Some manufacturers do not perform the tests on a routine basis but only as auxiliary tests. In some instances hospital clinical laboratories may run the tests. The levels of these chemicals in serum are important to end users and may also be used to assess lot-to-lot variability.
Endotoxin Levels
Although high endotoxin levels are not suitable for applications involving injectables, acceptable levels in bovine sera vary depending on the intended application. Some manufacturers may overlook the importance of low endotoxin levels in bovine sera used in cell culture applications. Endotoxin influences more than 30 biological activities. Some of these are macrophage activation, mitogenic stimulation, and induction of interferon and colony-stimulating factor (for some applications, these may be positive activities). Endotoxin can also lead to cytotoxicity by initiating complement activation. The most commonly used methods for endotoxin detection are the semiquantitative gel clot Limulus amebocyte lysate procedure and the quantitative kinetic chromogenic method described in Bacterial Endotoxins Test 85. For both the gel clot and the kinetic chromogenic assays, valid endotoxin assays require appropriate treatment by heat or dilution in order to avoid adverse effects of interfering substances in serum. Researchers should include a positive product control in each assay to confirm that any interference has been overcome by the heat or dilution treatment.
Osmolality
The osmolality test is designed to evaluate the electrolyte concentration in bovine serum. Chemicals that affect serum osmolality include sodium, chloride, bicarbonate, potassium, proteins, and glucose. Serum manufacturers should measure the osmolality of each serum batch to verify compliance with product specifications, using equipment calibrated with standards that are traceable to the National Institute of Standards and Technology. Osmolality and Osmolarity 785 describes how osmolality is determined by freezing-point depression of the bovine serum solution. Scientists use at least two standards to calibrate the instrument. The osmolality of each sample is calculated and related to the serum water content and is expressed as mOsmol/kg H2O.
Total Protein Level
The total protein level in serum is measured to verify animal age and compliance with product specifications. Biotechnology-Derived Articles—Total Protein Assay 1057 describes two procedures, the Biuret and Bradford methods, for determining protein concentration. The acceptable level of protein in serum should be assessed by the end user based on the intended application.
Cell Growth Properties
Each lot of serum should be tested for its ability to support in vitro growth of specific cell lines. Bovine sera are highly variable, and different lots may yield different results. Because of this variability, end users should characterize and standardize the cell lines that they will use for this type of testing. End users should design cell growth procedures that will help them check the efficacy of bovine serum in promoting cell growth. Serum manufacturers will benefit from monitoring growth promotion over several generations of subcultures to detect any evidence of cytotoxicity or changes in cell morphology. Different serum manufacturers use different cell types, and the growth studies and cell lines used by serum manufacturers also may differ from those applied by serum end users. When serum manufacturers evaluate the growth properties of a specific cell line in response to a specific lot of serum, they should take into account plating efficiency and/or growth promotion or some other functionality tests that qualify the serum lot for its intended use.
Plating efficiency at low cell density is a preferred method for analyzing the proliferative capacity and survival of single cells under optimal growth conditions. This procedure can reveal differences in the growth rate within the population and is capable of distinguishing between changes in growth rate (colony size) and cell survival (colony number). The growth kinetic is another important aspect in the design of cell-based experiments. Determining the growth curve of each cell line helps define optimal culture conditions, because variation in serum and other growth additives may influence growth parameters, which may affect the experimental outcome.
In the absence of specific tests designed for their particular products, serum users can refer to the functionality tests described in the proposed general chapter Fetal Bovine Serum—Quality Attributes and Functionality Tests 90 to determine whether a lot of serum is suitable for their application. This chapter provides guidance about how to perform growth promotion and plating efficiency tests.
In Vitro Cytotoxicity
Serum manufacturers should use an appropriate cell line for testing each lot of serum, and should perform growth studies through several subcultured generations to ensure that the serum has no cytotoxic effect on the cells. The choice of cell line depends on the intended use of serum. The cell growth and cytotoxicity assays should be performed on the final batch of serum after any viral inactivation step or any further processing.
CONCLUSION
Bovine serum is likely to remain an important component in the manufacture of many biologics, particularly those relying on cell culture. As with similar materials, bovine serum displays inherently variable quality. As a result, serum end users must establish suitable tests, procedures, and acceptance criteria for introduction of materials into a particular application process that uses serum. This may mean screening multiple lots of bovine serum to determine which lots meet the specification (see the section Characterization of Bovine Serum).
Manufacturers of therapeutic products using bovine serum are responsible for ensuring and documenting its quality and its impact on the quality, safety, and efficacy of the final product. In addition, it is important to ensure that each lot of serum performs in an equivalent manner during manufacturing. Serum can also interfere with final product purification; therefore it is important to understand the effect of bovine serum on the manufacturing process in order to understand the effect that various processes might have on the final product. Finally, risks can also be mitigated through the design of processes to include steps to adequately remove the bovine material through dilution, separation, or inactivation as well as the development of analytical assays to assess the bovine-derived residual content during processes and in the final therapeutic product. A number of sensitive assays can provide a quantitative means of detecting bovine material at picogram levels.
APPENDIX 1
Bovine sera and serum-related products used in the manufacture of biological products are regulated in the context of Requirements for Ingredients of Animal Origin Used for the Production of Biologics, 9 CFR 113.53. Currently, individual serum manufacturers perform detection studies to identify contaminating viruses. Because of the potential international market for serum, serum manufacturers need to be mindful of other regulatory requirements. Manufacturers can use the documents listed here as guidance for screening bovine sera for contamination by adventitious agents. Because of the risk carried by animal-derived serum products, serum manufacturers and end users should ensure that the country of origin of the material complies with applicable regulatory requirements. Although no cell performance assays currently demonstrate lack of BSE in serum, serum manufacturers must comply with the regulatory requirements of countries where the serum is sourced and marketed to ensure a minimal risk of infection with BSE/TSE.
Beyond relevant USP chapters referenced in this chapter, the following list of documents includes regulatory documents as well as best practices in product and process development, manufacturing, quality control, and quality assurance.
CFR
- 9 CFR 94.18 (CVB, 2001)
- 9 CFR 113.46
- 9 CFR 113.47
- 9 CFR 113.52
- 9 CFR 113.53
- 9 CFR 113.55
- 9 CFR 320
- 9 CFR 327.4
- 21 CFR 211 Subpart E
- 21 CFR 801.1
- 21 CFR 809.10
FDA
- FDA. Center for Biologics Evaluation and Research (CBER). 2000. Letter to manufacturers of biological products. Available at: http://www.fda.gov/BiologicsBloodVaccines/SafetyAvailability/ucm105877.htm
- Use of materials derived from cattle in medicinal products intended for use in humans and drugs intended for use in ruminants (Proposed Rule). Federal Register. 2007; 72(8): 1582–1619. Available at: http://www.reginfo.gov/public/
International Regulations and Guidance Documents
- CPMP/Biotechnology Working Party/EMEA (CPMP/BWP/EMEA). 1996. Note for guidance on virus validation studies: the design, contribution and interpretation of studies validating the inactivation and removal of viruses. Available at http://www.emea.europa.eu/pdfs/human/bwp/026895en.pdf.
- CPMP/BWP/EMEA. 2003. Note for guidance on the use of bovine serum in the manufacture of human biological medicinal products. Available at http://www.emea.europa.eu/pdfs/human/bwp/179302en.pdf.
- EMEA/CVMP/743/00-Rev.2 from the Committee for Veterinary Medicinal Products (CVMP). Revised guideline on requirements and controls applied to bovine serum used in the production of immunological veterinary medicinal products. Available at http://www.emea.europa.eu/pdfs/vet/iwp/074300en.pdf.
- EMEA/CPMP/BWP/1793/02, from the CPMP. Note for guidance on the use of bovine serum in the manufacture of human biological medicinal products. Available at http://www.emea.europa.eu/pdfs/human/bwp/179302en.pdf.
- CPMP/CVMP. Note for guidance on minimising the risk of transmitting animal spongiform encephalopathy agents via human and veterinary medicinal products. Available at http://www.emea.europa.eu/pdfs/human/bwp/TSE%20NFG%20410-rev2.pdf.
- World Health Organization (WHO), Office International des Epizooties. Terrestrial animal health code 2007. Available at http://www.oie.int/eng/normes/mcode/code2007/anc-en_summry.htm.
- WHO. 2006. WHO guidelines on tissue infectivity distribution in transmissible spongiform encephalopathies. http://www.who.int/bloodproducts/cs/TSEPUBLISHEDREPORT.pdf.
APPENDIX 2
Following is a general description of viruses that manufacturers can consider when testing bovine serum for the absence of adventitious agents. The list is intended only to provide general information. The list of required testing is described in this chapter in the section Viral Testing.
Akabane—
An insect-transmitted virus that causes congenital abnormalities of the central nervous system in ruminants. Disease due to Akabane virus has been recognized in Australia, Israel, Japan, and Korea. Antibodies to it have been found in a number of countries in Southeast Asia, the Middle East, and Africa. The disease affects fetuses of cattle, sheep, and goats. Asymptomatic infection has been demonstrated serologically in horses, buffalo, and deer (but not in humans or pigs) in endemic areas.
Bluetongue—
An infectious, noncontagious arthropod-borne viral disease primarily of domestic and wild ruminants. Infection with bluetongue virus is common worldwide but is usually subclinical or mild. Bluetongue virus is the type-species of the genus Orbivirus in the family Reoviridae. Worldwide, 24 serotypes have been identified, although not all serotypes exist in any one geographic area: e.g., only 5 serotypes (2, 10, 11, 13, and 17) have been reported in the U.S. Distribution throughout the world parallels the spatial and temporal distribution of vector species of Culicoides biting midges, which are the only significant natural transmitters of the virus.
Bovine adenovirus—
Associated with a wide spectrum of diseases. Bovine adenovirus type 3 is the serotype most often associated with bovine respiratory disease. Bovine adenoviruses are DNA viruses that have been separated into two genera: the Mastadenovirus, or mammalian adenoviruses, and the Aviadenovirus, or avian adenoviruses. Within the genus Mastadenovirus are numerous species-specific serotypes, nine of which have been identified in cattle. Epitheliotrophic adenoviruses have also been isolated from ruminants, and usually are clinically unapparent. Clinical disease is dictated by various factors, including the strain of virus, concurrent infection, stress, environmental conditions, and management practices.
Bovine herpesvirus 1 (BHV-1)—
Associated with several diseases in cattle, including infectious bovine rhinotracheitis, infectious pustular vulvovaginitis, balanoposthitis, conjunctivitis, abortion, encephalomyelitis, and mastitis. BHV-1 infections are widespread in the cattle population. In feedlot cattle the respiratory form is most common.
Bovine leukemia—
An exogenous C-type oncovirus in the family Retroviridae. Bovine leukemia is a viral disease of adult cattle characterized by neoplasia of lymphocytes and lymph nodes. Infection occurs by iatrogenic transfer of infected lymphocytes and is followed by a permanent antibody response. The prevalence of infection in a herd may be high, but only a few animals develop fatal lymphosarcoma. Infection is spread by contact with contaminated blood from an infected animal.
Bovine reovirus—
Double-stranded ribonucleic acid (RNA) (dsRNA) viruses with nonenveloped spherical virions 60–80 nm in diameter. They cause bovine respiratory diseases.
Bovine respiratory syncytial virus (BRSV)—
An RNA virus classified as a pneumovirus in the Paramyxovirus family. This virus was named for its characteristic cytopathic effect—the formation of syncytial cells. In addition to cattle, sheep and goats can also be infected by respiratory syncytial viruses. Human respiratory syncytial virus (HRSV) is an important respiratory pathogen in infants and young children. HRSV has antigenic subtypes, and preliminary evidence suggests the existence of antigenic subtypes of BRSV. BRSV is distributed worldwide, and the virus is indigenous in the cattle population. BRSV infections associated with respiratory disease occur predominantly in young beef and dairy cattle.
Bovine rotavirus—
A dsRNA spherical virion 60–80 nm in diameter without an envelope. It is the most common viral cause of diarrhea in calves and lambs.
Bovine viral diarrhea virus (BVDV)—
An RNA virus classified as a Pestivirus in the family Flaviviridae. BVDV can cross the placenta and appears to be capable of inducing immunosuppression, which allows the development of secondary bacterial pneumonia. BVDV has been reported to be the virus most frequently associated with multiple viral infections of the respiratory tract of calves.
Foot-and-mouth disease (FMD)—
A highly infectious viral disease of cattle, pigs, sheep, goats, buffalo, and artiodactyl wildlife species. In a susceptible population, morbidity approaches 100%. The disease is rarely fatal except in young animals. FMD is caused by an Aphthovirus of the family Picornaviridae. Seven immunologically distinct serotypes are known, and within each serotype exist a large number of strains that exhibit a spectrum of antigenic characteristics.
Parainfluenza-3 virus (PI-3)—
An RNA virus classified in the Paramyxovirus family. Although PI-3 is capable of causing disease, the virus usually is associated with mild to subclinical infection. The most important role of PI-3 is to serve as an initiator that can lead to the development of secondary bacterial pneumonia. Infections caused by PI-3 are common in cattle.
Parvovirus—
A relatively heat-stabile single-stranded DNA virus approximately 20 nm in diameter that has been recovered from cattle but under natural conditions is not known to cause disease.
Rabies—
An acute viral encephalomyelitis that principally affects carnivores and bats, although it can affect any mammal. Rabies is caused by Lyssaviruses in the Rhabdovirus family. Although they are usually confined to one major reservoir species in a given geographic area, spillover to other species is common.
Rinderpest—
A Morbillivirus, closely related to the viruses that cause canine distemper and measles. Strains may vary markedly in host range and virulence. Sera from recovered or vaccinated cattle cross-react with all strains in neutralization tests, but minor antigenic differences have been demonstrated. The virus is fragile and becomes rapidly inactivated by heat and light but remains viable for long periods in chilled or frozen tissues. Rinderpest is endemic in many countries in Asia and Africa. Historically, the virus has been widely distributed throughout Europe and Africa but to date has not established itself in North America, Central America, the Caribbean Islands, South America, Australia, or New Zealand. Rinderpest is included in the WHO’s Office International des Epizooties list of communicable diseases that have the potential for very serious and rapid spread, irrespective of national borders; that are of serious socioeconomic or public health consequence; and that are of major importance in the international trade of livestock and livestock products.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Fouad Atouf, Ph.D.
Senior Scientific Liaison 1-301-816-8365 |
(GCBA2010) General Chapters - Biological Analysis |
USP35–NF30 Page 449
Pharmacopeial Forum: Volume No. 35(3) Page 628