OVERVIEW
A number of glycoprotein drugs have been developed as a result of advances in biotechnology, and many naturally derived protein drugs possess complex glycan structures. Glycosylation, a posttranslational modification of these proteins, can play an important role in determining the function, pharmacokinetics, pharmacodynamics, stability, and immunogenicity of these agents. The two main types of protein glycosylation are N-glycosylation and O-glycosylation. Unlike transcription and translation, glycosylation is not a template-driven process; therefore variability in the glycosylation pattern of a protein can arise, caused by different sources or different manufacturing processes. Differences in this pattern are known to affect biological activity. Glycosylation patterns may therefore be an important set of attributes that arise in characterizing a candidate glycoprotein intended for therapeutic use and in ensuring its stability and quality.
The first part of this chapter provides a brief introduction to glycobiology and describes the complexity of glycan structures. The subsequent parts provide flow charts and a series of general analytical strategies that can be used to characterize glycoprotein glycans by means of the following:
- Direct analysis of glycoproteins; and
- Analysis of released nonderivatized or derivatized glycans by various methods of chromatographic and electrophoretic separation and mass spectrometry (MS).
Different approaches to analyzing monosaccharides are described at the end of the chapter.
For selected analytical methods, this chapter cross-references other USP chapters, particularly those relating to biotechnology-derived articles (see chapters Biotechnology-Derived Articles—Capillary Electrophoresis  1053
1053 , Biotechnology-Derived Articles—Isoelectric Focusing 1054, Biotechnology-Derived Articles—Peptide Mapping 1055, and Biotechnology-Derived Articles—Polyacrylamide Gel Electrophoresis 1056).
, Biotechnology-Derived Articles—Isoelectric Focusing 1054, Biotechnology-Derived Articles—Peptide Mapping 1055, and Biotechnology-Derived Articles—Polyacrylamide Gel Electrophoresis 1056).
PROTEIN GLYCOSYLATION
Most proteins in eukaryotic cells undergo glycosylation and other posttranslational modifications before being trafficked to lysosomes, becoming membrane bound at the cell surface, or being secreted. Glycosylation varies significantly from cell to cell, tissue to tissue, and species to species because of the varying expression of hundreds of glycosyltransferases and glycosidases located throughout the Golgi apparatus and endoplasmic reticulum (ER). Four main types of enzymatic glycosylation are found in proteins:
- N-Glycosylation, which involves the initial transfer of oligosaccharides to the nitrogen on the terminal amide group of asparagine and their subsequent processing and modification to a series of glycan chains;
- O-Glycosylation, which in general involves the initial transfer of monosaccharides to the hydroxyl groups of serine and threonine and subsequent elongation and branching of the saccharide chain by the addition of monosaccharides;
- Glycosylphosphatidylinositol (GPI) anchor, which is a glycolipid linked to the C-terminus of a protein; and
-
C-Glycosylation, which involves the formation of a carbon–carbon bond between the C2 carbon of the indole ring of tryptophan and the C1 carbon of an
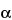 -mannopyranosyl residue.
-mannopyranosyl residue.
Any given protein may contain multiple N-, O-, or C-glycosylations, but not more than one GPI anchor. A nonenzymatic addition of saccharides, called glycation, can occur when proteins are mixed with reducing sugars via a complex series of reactions. The two protein glycosylation types that are generally of concern and that are analyzed in glycoprotein drug substances are N- and O-glycosylation. Each of these is discussed below.
N-Glycosylation
The biosynthesis of N-glycans in glycoproteins can be described as a four-step process:
- Lipid-linked glycan chain initiation and elongation;
- Transfer of oligosaccharide to the protein or nascent polypeptide chain;
- Processing of the N-glycan chain by removal of specific glucose and mannose residues; and
- Modification of the N-glycan chain by the addition of residues to the nonreducing ends of the glycan chain.
The consensus amino acid sequence for N-glycosylation is Asn–Xaa–Thr/Ser (where Xaa is any amino acid other than proline). Overall, only about two-thirds of all potential sequences, termed sequons, are glycosylated, and currently there is no method to predict which sequon will be glycosylated. The role of protein N-glycosylation is usually protein trafficking and secretion.
N-glycans can be categorized as high-mannose, hybrid, or complex, depending on the extent of processing (Figure 1). High-mannose structures (Appendix 1) lack galactose or N-acetylglucosamine (GlcNAc) residues in the antennae, branches at the distal end of the chain. In hybrid structures, both substituted GlcNAc residues and terminal mannose residues are present in the antennae, whereas complex structures have both 1,6- and 1,3-mannose residues substituted with GlcNAc moieties. Hybrid and complex glycans can exist with two or more branches, frequently termed antennae; such glycans are therefore often termed, for example, biantennary, triantennary, or tetraantennary. Both monoantennary and pentaantennary N-glycans are also known to exist. In complex glycans, antennae frequently carry terminal sialic acid (neuraminic acid) residues. Sialylation has been shown to have a great effect on both the pharmacokinetics and the pharmacodynamics of many therapeutic glycoproteins.
+'&img=../../images/v35300/c1084-f1-1susp34.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 1. Common types of N-glycans. For abbreviations, see Appendix 1.
O-Glycosylation
O-Glycan chains are built up sequentially via an initial GalNAc residue linked to serine, threonine, and tyrosine, as well as to the less common amino acids hydroxyproline and hydroxylysine. Multiple glycan core structures are known. The sequence and isomeric linkage of monosaccharides show greater variety than that in N-glycans, and at least eight different types have been identified (Figure 2). Although no consensus amino acid sequence for O-glycosylation has been determined, glycosylation is usually favored by the presence of proline one residue before or three residues after the glycosylation site and the absence of charged amino acid residues proximal to serine or threonine. The disaccharide unit N-acetyllactosamine, Gal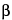 1,4GlcNAc, is the most common chain extension. Additional modifications, including terminal capping of Gal with sialic acid and fucosylation along the chain, are also frequent.
1,4GlcNAc, is the most common chain extension. Additional modifications, including terminal capping of Gal with sialic acid and fucosylation along the chain, are also frequent.
O-Glycosylation can occur in cluster form, the mucin type, which usually forms part of the cell surface extracellular matrix or secreted glycoproteins. Other O-glycosylation, such as O-GlcNAc, is found on many nucleocytoplasmic proteins; and O-Man–linked glycosylation is found in some muscular and neural glycoproteins and in yeast. O-Fuc– and O-Glc–linked glycosylation types are found on many epidermal growth factor-like proteins that are associated with the Notch signaling pathway.
+'&img=../../images/v35300/c1084-f2-1susp34.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 2. Common core structures of O-glycans (bold and underlined). Sugar abbreviations as in Appendix 1.
Glycan Heterogeneity
Not only the type of glycosylation (N- or O-linked), site occupancy, and the site of glycosylation can vary from glycoprotein to glycoprotein, but also the actual oligosaccharide structures (branching and linkages) can differ, even on the same site. This structural variation arises because glycosylation is a process that is not driven by a template. The glycosylation pattern at a given site depends on many factors, including cell-specific and growth-dependent availability of glycosyltransferases and exo-glycosidases found in the Golgi bodies and ER. Heterogeneity leads to different physical and biochemical properties and, therefore, also to functional diversity.
Host-Cell Expression Systems and Glycosylation
bacteria
Although both O- and N-glycosylation have been shown to occur in a variety of prokaryotes, Escherichia coli, the bacterium of choice for many therapeutic products, does not produce glycosylated proteins.
yeast
Yeast produces both N-glycosylated and O-glycosylated proteins. In yeast hypermannosylation with the N-glycan chain that contains more than 100 mannose residues can occur, but sialylation does not occur unless the organism is genetically modified. The development of recombinant strains of Pichia pastoris that contain inserted heterologous genes for various glycosylation enzymes has allowed the humanization of N-glycosylation pathways in this yeast. O-Glycosylation in yeast is also significantly different from that in mammalian cells. In contrast to mammalian cells, serine or threonine O-glycosylation is linked via mannose and often consists of linear chains of as many as six mannose residues.
insect cells
N-glycan chains of insect cells usually are of the high-mannose, trimannose or paucimannose, and truncated complex types (see Appendix 1 for definitions). Insect cells also produce glycoproteins bearing the Fuc1,3 residue linked to the proximal GlcNAc residue in the core chitobiose. This core fucose residue is a potent immunogen and allergen. O-Glycosylation in insect cells has not been well-studied, and although O-linked GalNAc–Ser(Thr) residues have been found, very few are processed further beyond the Gal1,3GalNAc–Ser(Thr) sequence. Sialic acid residues have not been found on proteins produced in insect cells.
plants and plant cells
Plant N-glycans contain mainly oligosaccharides of the oligomannose type, but also present are hybrid and truncated complex types of structures, with or without Xyl1,2 attached to the -linked mannose residue of the trimannosyl core and Fuc1,3 attached to the proximal GlcNAc residue of the core chitobiose. Both the Fuc and Xyl residues are immunogenic and have been shown to be part of the glyco-epitopes of several plant allergens. O-Glycosylation in plants has not been well studied but is known to consist predominantly of the addition of arabinogalactan chainsattached to hydroxyproline, threonine, and serine residues that are located in the plant cell wall or on the outer surface of the plasma cell membrane. These glycans are immunogenic.
animal cells
The majority of glycosylated therapeutic proteins are produced in continuous animal cell lines. Chinese hamster ovary (CHO), baby hamster kidney (BHK), human embryonic kidney (HEK), and mouse myeloma (SP2/0 or NS0) cells have all been employed. These animal cells generally produce proteins with humanlike glycosylation. Although there are several differences in glycosylation between rodent and human cells, such as the presence of N-glycolylneuraminic acid not found in humans, CHO cells have become a workhorse of the biotechnology industry.
GLYCAN ANALYSIS FOR GLYCOSYLATED BIOLOGICAL DRUGS
Glycosylation of proteins may affect biological activity, either directly or indirectly, and variability in glycosylation arises not only from cellular diversity but also from the manufacturing process. The glycosylation pattern thus may be important as a part of characterization studies in assuring process consistency and may be also be important in ensuring the consistent quality of a biological drug product after market access. Appropriately characterized reference materials are needed in order to support biological and physicochemical testing of production batches to ensure batch-to-batch consistency. Glycosylation analysis may be appropriate for the following:
- Characterizing the structure and stability of novel products and their stability to processing steps and storage;
- Batch release testing and in process control testing; and
- Assessing comparability between products (e.g., when one or more process changes have been made).
An understanding of the relationship between glycan structure and biological function underpins decisions about the information required at each development stage. For biological/biotechnological drug substances, the characterization criteria and specifications for batch release are generally set forth in the guidelines ICH Q6B, Test Procedures and Acceptance Criteria for Biotechnological/Biological Products;1 and ICH Q5E, Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process.2 Numerous approaches and methodologies are applied for glycan mapping. This variety is a consequence of the diversity and complexity of glycan structures and the available technology and detection systems.
Because of the diversity and complexity of glycan structures and the increasing availability and improvement of various detection systems and technology, analytical methods are wide ranging. Different methods that support step-by-step procedures depend on the glycoproteins, the availability of equipment, the expertise of individual scientists and groups, and the information required. The two most studied types of protein glycosylation that affect bioactivity are N- and O-glycosylation.
Glycan analysis can serve in different applications; the most important are general product characterization, process validation, comparability evaluation, stability testing, monitoring manufacturing process consistency, and release testing. The selection of the analytical techniques and their applications in product development and routine manufacturing depend on many factors, such as the complexity of the glycoprotein, the understanding of the relationships between glycosylation and safety and efficacy, and the overall design of the strategy for manufacturing process control. For example, even when the biological relevance of glycosylation is not certain, control of glycosylation could be considered as a measure of manufacturing consistency.
+'&img=../../images/v35300/c1084-f3a-1susp34.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 3A. Flow diagram assisting in the choice of options for glycan analysis.
+'&img=../../images/v35300/c1084-f3b-1susp34.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 3B. Overview of glycan analysis techniques and equipment employed.
CHOICE OF GLYCAN ANALYSIS FOR CHARACTERIZATION AND SPECIFICATION OF GLYCOSYLATED BIOLOGICAL DRUGS
Analysis of Intact Glycoprotein
The most direct mode of analysis is direct study of the intact molecule. This mode provides information about the glycosylation profile of the glycoprotein. However, this approach provides limited information when the molecule is large and contains multiple glycosylation sites. One of the most important glycosylation factors defining biological activity is the degree of sialylation, which often determines the half-life of glycoproteins in circulation. This makes ionic-charge–based electrophoresis and ion-exchange chromatography obvious choices of technique. Nearly all types of gel electrophoresis have been used to probe protein glycosylation, including polyacrylamide gel electrophoresis (PAGE), (see Biotechnology-Derived Articles—Polyacrylamide Gel Electrophoresis 1056) and isoelectric focusing (IEF), (see Biotechnology-Derived Articles—Isoelectric Focusing 1054). Similarly, capillary electrophoresis (CE), (see Biotechnology-Derived Articles—Capillary Electrophoresis 1053) has also been found suitable. Strong anion-exchange chromatography has been used for the same purpose, but the resolution is often inferior to that of IEF and CE. Direct mass spectrometry (MS) is another option for the analysis of posttranslational modification. Along with ongoing improvements in the resolution of MS, more and more complex glycoproteins become accessible for direct characterization by this method.
Analysis of Glycopeptides
Analysis of glycopeptides provides information about site-specific glycosylation properties, the degree of occupancy, and oligosaccharide structures. Site-specific glycosylation can be affected by cell culture process conditions. Therefore, if a known glycosylation site is critical, manufacturers must monitor site-specific glycan structures. The typical approach is first to generate glycopeptides by protease digestion and to separate them by, for example, RP (reversed-phase) HPLC (see Biotechnology-Derived Articles—Peptide Mapping 1055). Subsequently the separated glycopeptides can be further characterized individually by, e.g., direct analysis using MS, or deglycosylation and subsequent glycan profiling, as described below in the section Profiling of Cleaved Oligosaccharides.
Direct identification of the mixture of glycopeptides and nonglycosylated peptides by MS is limited by masking effects (ion suppression) of peptide signals on glycopeptide signals. One approach to overcoming this effect is to separate peptides and glycopeptides before analysis by MS, e.g., by offline coupling (matrix-assisted laser desorption ionization [MALDI]) or online coupling (electrospray ionization [ESI]). MS analysis of glycopeptides plays an important role in the characterization of O-glycans because these glycans are not always released quantitatively and because, as a result of their smaller size, they are more amenable to characterization by MS as glycopeptides.
The use of CE for high-resolution separation may also be appropriate, especially for the analysis of sialylation.
Profiling of Cleaved Oligosaccharides
Profiling of total glycans cleaved from glycoprotein is the most common approach for the characterization of glycoproteins. It provides a way to obtain information about the various populations of glycans present on the protein. The degree of sialylation can also be addressed at this stage. Depending on the chosen method, prior derivatization/labeling may be needed to allow the detection of the glycans. Many protocols are available, and most of the steps in the analysis are well established. The possible drawback with such flexibility is the lack of consensus about which methods to choose under which circumstances; because of the variety of analytical techniques, comparison of resultsobtained by different platforms may not always be possible. So far, the majority of the work has been done on N-glycosylation, because of the following factors:
- N-glycans usually are more clinically relevant in biologicals than O-glycans; or
- The release of N-glycans, either by chemical means (hydrazine) or by enzymes (endoglycosidases and peptide N-glycosidase [PNGase] F), is more straightforward than is the release of O-glycans.
deglycosylation
The approach used for the release of glycans depends on the glycoprotein under test. The cleavage agent is chosen according to the type of cleavage needed and the level of information required. Enzymatic or chemical cleavage may be used. Table 1 gives a nonexhaustive list of enzymatic cleavage agents and their specificity. Digestion efficiency generally depends on the accessibility of the glycans on the protein, and hence the protein should be denatured to maximize glycosylation site exposure unless analysts want to distinguish between surface and buried glycans. Chemical cleavage agents can also be used, e.g., hydrazine or alkaline borohydride for -elimination of O-linked glycans.
Table 1. Examples of Enzymatic Cleavage Agents
| Agent | Specificity |
|---|---|
| N-linked glycan release | |
| Peptide-N4-(N-acetyl- |
Hydrolysis of peptide-N4-(N-acetyl- |
| Peptide N-glycosidase F (PNGase F) | Release of N-glycan chain but no release of N-glyc an chain containing ( |
| Peptide N-glycosidase A (PNGase A) | Release of N-glycan chain containing ( |
| Mannosyl-glycoprotein endo- |
Endohydrolysis of the N,N'-diacetylchitobiosyl unit in high-mannose glycopeptides/glycoproteins containing the –[Man(GlcNAc)2]Asn structure |
| Endo- |
Release of high-mannose, hybrid, and complex oligosaccharides |
| Endo- |
Release of high-mannose and hybrid oligosaccharides |
| O-linked glycan release | |
| Glycopeptide |
Hydrolysis of terminal d-galactosyl-N-acetyl- |
|
*
This enzyme has limited usage because of its high substrate specificity.
|
|
Chemical or Enzymatic Release of N-Glycans
—PNGase F (Flavobacterium meningosepticum) is the enzyme of choice for the release of N-glycans for most glycoproteins except for some insect cell and plant glycoproteins that may contain a Fuc1,3 linked to the chitobiosyl core. N-Glycan chains having this structure can be cleaved from the glycopeptide only by the almond enzyme, PNGase A. Chemical release by anhydrous hydrazine is much less common, mainly because of the limited availability of the reagent, which is considered a hazardous chemical. In addition, hydrazinolysis produces de–N-acetylated N-glycans.
Chemical or Enzymatic Release of O-Glycans
—Currently only one enzyme, O-glycanase from Diplococcus pneumoniae, is available to release O-glycans, and this enzyme has a limited usage because of its high substrate specificity: it cleaves only Gal1,3GalNAc1-Ser/Thr. In addition, no ideal chemical procedure is available; but Ser- and Thr-linked O-linked glycan can usually be released by the reductive alkali-catalyzed -elimination reaction (alkaline borohydride reaction), in which the released glycans are reduced as soon as they are cleaved in order to prevent formation of degradation products due to peeling. However, this reaction is not specific, and in the reaction, approximately 10%–20% of N-glycans are generally known to be released as well. The released glycans lack a reducing group used for the attachment of fluorescent labels by reductive amination. Fortunately, with advances in sensitive MS, direct identification of reduced glycans is possible. Relatively good quality reducing O-glycans can be obtained by alkali-catalyzed -elimination using primary amines such as ethylamine and hydrazine. However, both reagents have the potential to produce peeled degradation products. Furthermore, O-glycan release by ethylamine is not quantitative. Hydrazine, although it may be better for use than ethylamine, requires strict control of reaction conditions and handling and does not have a commercial source in Europe.
Separation of Cleaved Glycans Without Fluorescent Labeling
—N-Glycans can also be resolved by HPAEC high-pH anion-exchange chromatography with pulsed amperometric detection (PAD; see Chromatography 621), which shows high sensitivity, can also separate some isomers, and affords the ability to directly detect native glycans without labels or tags. However, LC/MS for this separation approach is challenging because this HPAEC system uses high-pH and high-salt mobile phases that interfere with ionization of glycans. In addition, absolute quantification of the glycan is only possible if the individual PAD response factors for the different glycan structures are known, e.g., if an appropriate oligosaccharide reference library is available. Porous graphitic carbon (PGC) chromatography can also be used to separate glycans, and this method adds an orthogonal selectivity compared to other columns. A PGC-electrospray-ionization-MS approach also can be applied for direct glycan analysis.
MALDI/ESI-MS is a powerful method for the analysis of glycan mixtures either in the native or derivatized form. Permethylation of released glycans is a common method for direct analysis using MALDI/ESI-MS especially for sialylated glycans.
labeling of glycans to increase detection sensitivity and/or to modify their physicochemical properties
Chemical derivatization is the most commonly used method for labeling glycans at their reducing end by reductive amination. One fluorescent label can be attached to each mono- and oligosaccharide, which facilitates determination of molar quantities. Table 2 illustrates the most common examples of fluorescent labels and their most common uses.
Table 2. Examples of Fluorescent Labels
| Name | Acronym | Structure | Analytical Technique |
|---|---|---|---|
| 2-Aminobenzoic acid | 2-AA |
+'&img=../../images/v35300/c1084-structure1.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
|
HPLC |
| 2-Aminobenzamide | 2-AB |
+'&img=../../images/v35300/c1084-structure2.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
|
HPLC MS |
| 2-Aminopyridine | 2-AP |
+'&img=../../images/v35300/c1084-structure3.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
|
HPLC |
| Trisodium 8-aminopyrene-1,3, 6-trisulfonic salt |
APTS |
+'&img=../../images/v35300/c1084-structure4.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
|
CE |
n-glycan profiling
Released glycans can be analyzed or profiled by chromatographic, electrophoretic, or MS procedures and, in general, by a combination of these. The choice of method can be grouped according to the nature of the glycans and level of information required. Analysis of glycans provides information about the various populations of glycans present on the protein (high-mannose, hybrid, complex).
Profiling of Glycans by HPLC and/or by Electrophoresis and MS
—Profiling of fluorescent-tag–labeled glycans by HPLC has become the most common approach. One label can be attached to every single mono- and oligosaccharide by reductive amination at their reducing end, which facilitates determination of molar quantities. With the appropriate label, glycans can be profiled with high sensitivity using reversed-phase, normal-phase, and anion-exchange HPLC (see Chromatography 621). Routinely, analysts use a combination of these methods in order to increase separation resolution and to better differentiate glycan structures. The accuracy of the glycan identification can be validated by means of glycan standards and/or by coupling the HPLC system with MS. Thus, anion-exchange, normal-phase, and reversed-phase HPLC–ESI–MS–MS form powerful combinations; and in-line analysis, if possible, may provide both relative quantitative profiling and information on glycan structure in a single run. Peak identification through retention time is acceptable if their identities have been previously validated by complementary methods and peak homogeneity can be assured.
The degree of sialylation of glycan chains can be a crucial factor for clinical efficacy, because sialylation often defines the half-life of the molecule in vivo. Anion-exchange HPLC is the simplest method for its determination, and glycan structures based on charge can then be identified by MS. Desalting of each fraction is required before MS if the ionization interface is designed for low-salt-containing sample flows only.
High-resolution separation systems such as CE have been used to identify glycan structures without MS when well characterized standards are used for comparison. The development of an online CE–MS system has further increased the power of glycan analysis using this approach.
Structural Identification by Micro-Enzyme Sequencing and Mass Spectrometry
—Traditionally, when detailed structural information is required, the analysis is usually performed using micro-enzyme sequencing. This procedure is highly dependent on the specificity and quality of the enzymes used. Recently, tandem MS has been used more regularly to confirm, determine, and sequence known and novel glycan structures; this method is feasible especially when glycans are released from well-known glycoproteins and production sources.
Monosaccharide Analysis
Different quantitative monosaccharide assays are carried out for a number of purposes. In the glycoprotein field, they provide information about the relative amounts of saccharide in a glycoprotein and about the degree of sialylation of a glycoprotein; and by the measurement of monosaccharide composition, they provide some information about the structure of the glycan chains present.
The simplest assays used are colorimetric tests to demonstrate that the product is glycosylated and to quantify the total amount of saccharide present in the product. These have poor specificity between different types of sugar residues.
Assays of monosaccharide composition are generally simpler to perform than is oligosaccharide profiling, but they provide less information. The most widely used assay is quantification of sialic acid content, because loss of sialylation and exposure of terminal Gal residues may lead to faster clearance of the glycoprotein from the circulation.
The assays can be divided into two types: (1) those that provide compositional information about the intact sample without prior degradation; and (2) others, principally chromatographic, that require hydrolysis of the saccharide chains before analysis and generate quantitative information about several different monosaccharide species simultaneously. In general, the former are colorimetric and the latter are chromatographic. The hydrolysis step is a significant source of assay variability and may require careful optimization for specific samples.
The presence of certain monosaccharides is diagnostic of specific glycan structures. For example, observation of GalNAc is usually a marker for the presence of O-linked glycan chains, and fucose denotes the presence of specific types of chains. As a consequence of the limited diversity of monosaccharide residues present in glycoprotein glycans, accurate quantification of Man, Gal, or GlcNAc residues is required in order to distinguish between large numbers of structurally diverse glycans. The monosaccharide N-glycolylneuraminic acid (Neu5Gc) is not produced in humans and is generally regarded as an unwelcome and potentially immunogenic component of biopharmaceutical products.
sample preparation
Glycoprotein samples for monosaccharide analysis should be free of salts, excipients, and other carrier materials (low molecular weight sugars are often used as excipients for biopharmaceuticals). This can be achieved by a number of methods, including the following:
- Dialysis against water or a volatile buffer, using an appropriate membrane, and lyophilization;
- HPLC on an appropriate gel-permeation column eluted with water or a volatile buffer, monitored by UV absorbance or refractive index, and followed by lyophilization of the sample; or
- Sample trapping on a conventional RP-SPE cartridge such as a C18 or C8 SPE system, followed by washing away of salts and excipients and elution of the required sample.
quantification
The common method for quantification of neutral sugars in glycoproteins depends on the color generated by heating glycans or glycoproteins in the presence of aqueous phenol in concentrated sulfuric acid. In many cases, the heat required for this reaction is generated by addition of concentrated sulfuric acid to the glycoprotein–phenol mixture in water. Rapid and efficient mixing of the solutions is critical for consistent results. Quantitative results are obtained by the simultaneous analysis of standards to generate a standard curve of absorbance against amount of saccharide and/or against a reference sample of the product under analysis.
hydrolysis procedures for polysaccharides and glycoprotein glycan chains
Chromatographic methods for the identification and quantification of monosaccharide components require hydrolysis of the sample before analysis. Appropriate sample preparation is required because excipients or process-related impurities may be saccharides, and residual salts may interfere with the hydrolysis or the subsequent chromatographic separation or with fluorophore labeling. Sialic acid residues can be released either by mild acid hydrolysis or by enzymatic treatment, which leaves other sugar residues attached to the peptide backbone. Quantification of the amount of saccharide present is based on addition of an internal standard before or after hydrolysis. The most commonly used standard for sialic acid analysis by HPAEC is 3-deoxy-d-glycero-d-galacto-2-nonulosonic acid (KDN), and 2-deoxyglucose is widely used for neutral sugars. Both of these sugars are acid labile and should be added after the hydrolysis step. Accurate quantification depends both on stoichiometric hydrolysis and a lack of degradation of the monosaccharide products during hydrolysis.
determination of total sialic acids
Sialic acids occur in bacterial polysaccharides and glycoproteins generally as N-acetyl and N-glycolyl derivatives of neuraminic acid (Neu5Ac and Neu5Gc). The sialic acids can be determined together with other monosaccharides by a procedure that includes acid hydrolysis to liberate constituent monosaccharides, followed by HPLC using an appropriate standard mixture. Alternatively, total sialic acid content can be determined by colorimetric procedures without the need for hydrolysis. One method, commonly referred to as the Warren method, is based on the reaction of thiobarbituric acid with the product of periodate oxidation of neuraminic acid released in situ from the glycoprotein. Alternatively, the color can be generated by the reaction of resorcinol with neuraminic acid. For accurate quantification, include a reference standard sample is included in each measurement.
Selective Release of Sialic Acids
—Mild acid hydrolysis or enzymatic digestion can be used to selectively release sialic acid from glycoprotein glycan chains for quantification by chromatographic methods and for quantification of unwelcome forms such as Neu5Gc. More aggressive acid conditions are required in order to release neutral and amino sugars before chromatographic analysis. The protocol must be optimized with respect to yield and saccharide degradation for each protein to be analyzed.
Neuraminidase Digestion for the Release of Sialic Acid from Intact Glycoproteins
—Several types of neuraminidases have been isolated and studied; the enzyme derived from Clostridium perfringens is the one most commonly used for the enzymatic release of sialic acids from glycoproteins. Recombinant enzyme is available from commercial suppliers. Other enzymes with different specificities are available and can be used to distinguish different types of linkages. Hydrolysis conditions should be optimized for each product, because kinetic parameters for different linkages and for Neu5Ac and Neu5Gc may differ. Selective removal of Neu5Ac2®,3-linked and Neu5Ac2®,6- from cleaved glycans is a convenient means of defining linkages. For quantitative analyses, a known quantity of a suitable internal standard, often 2-deoxyglucose, is added after hydrolysis and removal of the acid.
separation and quantitation of unlabeled monosaccharides
Essentially the only method used for the simultaneous identification and quantification of unlabeled monosaccharide in hydrolysates is HPAEC-PAD. HPAEC-PAD methods are also applicable to oligosaccharide separations, and a single instrumental approach can be used for both applications.
For the general principles and components of chromatography, see Chromatography 621. HPAEC-PAD facilitates analysis of monosaccharides and all classes of oligosaccharides without derivatization. Carbohydrates, because they are polyhydric compounds, are weak acids that have pKa values of 12–14, and at high pH even neutral carbohydrates are ionized and can be separated as oxyanions by ion-exchange chromatography. Although separations can be performed on alkali-stable porous polystyrene–divinylbenzene anion exchangers, carbohydrates tend to exhibit broad peaks as a result of mass transfer problems. In microbead pellicular anion-exchange column packings, small functionalized latex beads (<0.1-µm diameter) are attached to larger (<10-µm diameter) uniform nonporous beads. The carbohydrate analyte interacts with the functional groups at the surface of the latex microbeads, eliminating diffusion into and out of pores and the associated peak broadening.
PAD is the method of choice for the detection of carbohydrates in HPAEC because it relies on the high-pH solutions that HPAEC provides by default. Amperometric detection measures the current, or charge, resulting from the oxidation or reduction of analyte molecules at the surface of a working electrode. Electrons are transferred from the electroactive analyte to the electrode during oxidation reactions and in the opposite direction during reduction reactions. This process allows sensitive and highly selective detection of analytes that can be oxidized or reduced, but interfering species that are not electroactive remain undetected. Carbohydrates are easily oxidized at gold and platinum electrodes at high pH, and the current generated is proportional to the carbohydrate concentration.
A typical amperometric detection system contains a working electrode and a reference electrode. Gold electrodes are most common for carbohydrate analysis, but oxidation products poison the electrode surface and inhibit further oxidation. Maintaining a stable, active electrode surface is accomplished by cyclical pulsing between high positive and negative potentials. This timed series of different potentials is referred to as a waveform, and repeated application of a waveform is the basis of pulsed amperometry. Different waveforms are used for different HPAEC-PAD applications and for different working electrodes: disposable gold electrodes require the use of fast, quadruple waveforms, but other gold electrodes allow a wider range of waveforms to be used without damaging the electrode surface. Disposable electrodes and fast waveforms were introduced to minimize the influence of electrode recess on the sensitivity and precision of quantitative monosaccharide applications.
fluorophore labeling of monosaccharides before separation and quantification
An alternative approach to the identification and quantification of monosaccharides present in a hydrolysate is to modify the monosaccharides by reductive amination with an easily detected fluorophore label that allows high-sensitivity detection and improves the chromatographic separation of monosaccharides. Essentially standard HPLC equipment can be used and, because the same labeling approaches are applicable to cleaved oligosaccharides, a consistent analytical approach can be applied. Fluorophore labeling has been much less widely used than HPAEC-PAD for monosaccharide identification and quantification. Labeling of sialic acid derivatives is usually undertaken with 1,2-phenylenediamine (or DMB, the 4,5-methylenedioxy derivatives), and the resulting products are separated on a C-18 column and using fluorescence detection.
CONCLUSION
Because of the complexity of glycoprotein glycan structures and their inherent variation during production processes, manufacturers are generally required by means of characterization studies to develop criteria for the control of the glycosylation pattern of a biological drug substance when glycosylation occurs, as well as to develop the level of information required at each stage of production and at batch release. Then analytical procedures can be derived in a manner that provides information relevant to fulfilling quality requirements. In general, a combination of approaches and techniques is needed, and more detailed glycan structural analysis at early drug development stages is required. Validation considerations are central as method development and product knowledge progress.
APPENDIX 1
Abbreviations
| Fuc | l-Fucose |
| Gal | d-Galactose |
| GalNAc | N-Acetyl-d-galactosamine |
| Glc | d-Glucose |
| GlcNAc | N-Acetyl-d-glucosamine |
| Man | d-Mannose |
| Neu5Ac | N-Acetylneuraminic acid |
| Neu5Gc | N-Glycolylneuraminic acid |
| Xyl | d-Xylose |
Additional Definitions
High mannose—Glycan chains containing two core GlcNAc residues and between five and nine Man residues, and lacking Gal, GlcNAc, or Neu5Ac residues in the antennae. Such chains are typically found in mammalian glycans.
Hypermannosylation—(i) Addition of Man residues to high mannose chains creating chains with large numbers of Man residues, and (ii) O-Man linked glycan chains with multiple Man residues synthesized by yeast.
Paucimannose—Glycan chains containing two core GlcNAc residues between two and four Man residues. Core-linked Fuc1,3 and/or Fuc1,6 residues may be present.
Oligomannose—Used here as a generic term to include high mannose, paucimannose, and N-linked hyper-mannosylated chains.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Tina S. Morris, Ph.D.
Vice President, Biologics & Biotechnology 1-301-816-8397 |
(GCBA2010) General Chapters - Biological Analysis |
USP35–NF30 Page 649
Pharmacopeial Forum: Volume No. 36(2) Page 486