



Add the following:
INTRODUCTION
Ultraviolet-visible (UV-Vis) spectra are derived when the interaction between incident radiation and the electron cloud in a chromophore results in an electronic transition involving the promotion of one or more of the outer shell or the bonding electrons from a ground state into a state of higher energy. The UV and visible spectral bands of substances generally are broad and do not possess a high degree of specificity for compound identification. Nevertheless, they are suitable for quantitative assays and, for many substances, are useful as an additional means of identification.
In the Beer–Lambert law the absorbance (A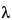 ) of a solution at given wavelength, , is defined as the logarithm to base 10 of the reciprocal of the transmittance (T):
) of a solution at given wavelength, , is defined as the logarithm to base 10 of the reciprocal of the transmittance (T):
+'&img=../../images/v38332/c857-eq1-pf375.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
I
I
In the absence of any other physical or chemical factors, A is proportional to path length, b, through which the radiation passes, and to the concentration, c, of the substance in the solution in accordance with the following:
A =  cb
cb
c = solute concentration (mol/L)
b = path length (cm)
If the concentration, c, is expressed in g/L, the constant becomes a, which is called the absorptivity.
The expression
+'&img=../../images/v38332/c857-eq2-pf375.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
which represents the specific absorbance of a dissolved substance, refers to the absorbance of a 10-g/L solution (1% m/v) in a 1-cm cell measured at a defined wavelength so that:
+'&img=../../images/v38332/c857-eq3-pf375.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
M = molar concentration of the solution
When solutions are observed in 1-cm cells, concentrations of about 10 µg of the specimen per mL often will produce absorbances of 0.2–0.8 in the UV or visible region.
For discussion of the theory and principles of measurements, see Ultraviolet-Visible Spectroscopy—Theory and Practice  1857
1857 , a general information chapter that is not a mandatory resource.
, a general information chapter that is not a mandatory resource.
QUALIFICATION OF UV-VIS SPECTROPHOTOMETERS
The suitability of a specific instrument for a given procedure is ensured by a stepwise life cycle evaluation for the desired application from selection to instrument retirement: design qualification (DQ); installation qualification (IQ); an initial performance-to-specification qualification, also known as operational qualification (OQ); and an ongoing performance qualification (PQ). For more details, see Analytical Instrument Qualification 1058.
The purpose of this chapter is to provide test methodologies and acceptance criteria to ensure that the instrument is suitable for its intended use (OQ), and that it will continue to function properly over extended time periods as part of PQ. As with any spectrometric device, a UV-Vis spectrophotometer must be qualified for both wavelength (x-axis) and photometric (y-axis, or signal axis) accuracy and precision, and the fundamental parameters of stray light and resolution must be established. OQ is carried out across the operational ranges required within the laboratory for both the absorbance and wavelength scales.
Installation Qualification
The IQ requirements provide evidence that the hardware and software are properly installed in the desired location.
Operational Qualification
Acceptance criteria for critical instrument parameters that establish “fitness for purpose” are verified during IQ and OQ. Specifications for particular instruments and applications can vary depending on the analytical procedure used and the desired accuracy of the final result. Instrument vendors often have samples and test parameters available as part of the IQ/OQ package.
Wherever possible in the procedures detailed as follows, certified reference materials (CRMs) are to be used in preference to laboratory-prepared solutions. These CRMs should be obtained from a recognized accredited source and include independently verified traceable value assignments with associated calculated uncertainty. CRMs must be kept clean and free from dust. Recertification should be performed periodically to maintain the validity of the certification.
Control of Wavelengths
Ensure that the accuracy of the wavelength axis (x-axis) over the intended operational range is correct within acceptable limits.
For non-diode array instruments, wavelength accuracy and precision are determined over the operational range using at least six replicate measurements. For wavelength accuracy, the difference of the mean measured value to the certified value of the CRM must be within ±1 nm in the UV region (200–400 nm), and in the visible region (400–700 nm) must be within ±2 nm. For wavelength precision, the standard deviation of the mean must not exceed 0.5 nm. For diode array instruments, only one wavelength accuracy measurement is required, and no precision determination needs to be performed. The difference between the certified and measured value of the CRM must not exceed ±1 nm in the UV region (200–400 nm), and in the visible region (400–700 nm) must not exceed ±2 nm.
atomic line spectra
This procedure is described as the primary application because the emission lines produced from a discharge lamp are characteristic of the source element and, as a fundamental physical standard, these wavelengths have been measured with an uncertainty of NMT ±0.01 nm. In solution spectrometry, the wavelength accuracy required rarely exceeds 0.5 nm. For these reasons, the atomic line standard values are cited without uncertainty. The lamp needs to be placed at the source position in the spectrophotometer; thus, it can be used only in spectrophotometers that can be operated in a single-beam intensity mode and practically should be implemented only on a system designed to accommodate these sources, e.g., as an accessory.
A commonly employed low-pressure mercury lamp has a number of intense lines that cover a large part of the UV and visible spectra. Two deuterium lines from the source at 486.0 and 656.1 nm often are used by manufacturers as an internal calibration check and can be used for diagnostic purposes (Table 1).1
Table 1. Recommended Atomic Lines from Low-Pressure Mercury and Deuterium Lamps for Wavelength Calibration Purposes
| Element |
nm |
|---|---|
| Hg |
253.7 |
| Hg |
296.7 |
| Hg |
365.0 |
| Hg |
404.7 |
| Hg |
435.8 |
| D2
|
486.0 |
| Hg |
546.1 |
| Hg |
577.0 |
| Hg |
579.1 |
| D2
|
656.1 |
rare earth oxide solutions
This procedure uses solutions of rare earth oxides prepared by dissolution in acid media. The most frequently used is holmium oxide in perchloric acid. Holmium oxide solution has been internationally accepted as an intrinsic wavelength standard, and suitable CRMs are available commercially.2 The observed peak maxima are determined using the normal scan mode on the spectrophotometer. The peak maxima for a 4% m/v solution of holmium oxide in perchloric acid at 1.0-nm spectral bandwidth and a path length of 1 cm are shown in Table 2.3
Table 2. Recommended Peak Maxima from a 4% Solution of Holmium Oxide in Perchloric Acid for Wavelength Calibration Purposes
| nm |
|---|
| 241.1 |
| 249.9 |
| 278.1 |
| 287.2 |
| 333.5 |
| 345.4 |
| 361.3 |
| 385.6 |
| 416.3 |
| 451.4 |
| 467.8 |
| 485.2 |
| 536.6 |
| 640.5 |
If the operational range of the spectrophotometer lies outside the range 240–650 nm, other certified rare earth oxides or other solutions can be used if they are traceable to a national or international standard. Didymium (a mixture of neodymium and praseodymium) is available as a traceable standard both in solution and as a glass. Didymium is similar in preparation to the holmium materials and has useful peak characteristics in the 730–870 nm region. Useful peaks are found in the didymium solution at approximately 731.6, 740.0, 794.1, 799.0, and 864.4 nm.
rare earth glasses
This procedure uses glasses manufactured by fusing the appropriate rare earth oxide in a base glass matrix. The most frequently used is holmium, for which the reference wavelengths have been well defined. Although manufacturing can cause batch variation in these glasses, traceable CRMs are commercially available and can be used. Typical values for a holmium glass using a 1.0-nm spectral bandwidth are the following: 241.5, 279.2, 287.5, 333.8, 360.9, 418.8, 445.8, 453.7, 460.2, 536.5, and 637.7 nm.
Control of Absorbance
To establish the transmittance accuracy, precision, and linearity of a given system, it is necessary to verify the absorbance accuracy of a system over its intended operational range by using the following procedures as appropriate for the wavelength and absorbance ranges required.
acidic potassium dichromate solutions in 0.001 m perchloric acid
In the 0–200 mg/L range, potassium dichromate solutions provide reference values of up to 3 absorbance units at one of the certified values of 235, 257, 313, or 350 nm. These solutions are available as CRMs or can be prepared according to NIST from SRM 935a. Using potassium dichromate solutions, the absorbance accuracy must be ±1%A (for values above 1.0A) or ±0.010A (for values below 1.0A), whichever is larger. The absorbance precision can be determined as the standard deviation of at least six replicate measurements at two or more absorbance levels over the operational range. The standard deviation must not exceed ±0.5%A (for values above 1.0A) or ±0.005A (for values below 1.0A), whichever is larger.
neutral-density glass filters
These gray glass filters are manufactured from doped glass and have a nominally flat spectrum in the region of the calibration wavelengths. They provide reference values of up to 3 absorbance units at the certified values of 440, 465, 546.1, 590, and 635 nm. These filters are available as CRMs that are traceable to NIST SRM 930e, 1930, and 2930. Other certified standard solutions or optical filters can be used if they are traceable to a national or international standard. Using gray glass filters, the absorbance accuracy must be ±0.8%A (for values above 1.0A) or ±0.0080A (for values below 1.0A), whichever is larger. The absorbance precision can be determined as the standard deviation of at least six replicate measurements at two or more absorbance levels over the operational range. The standard deviation must not exceed ±0.5%A (for values above 1.0A) or ±0.005A (for values below 1.0A), whichever is larger.
Limit of Stray Light (Stray Radiant Energy)
Although the measurement of absorbance or transmittance is a ratio measurement of intensities and therefore theoretically is independent of monochromatic source intensity, practical measurements are affected by the presence of unwanted radiation called “stray radiant energy” or “stray light”. In addition, the adverse effect of stray light increases with aging of optical components and lamps in a spectrophotometer. The effects are greater at the extremes of detector and lamp operational ranges. Analysts must monitor the level of stray light at appropriate wavelength(s) as part of PQ. Stray light can be detected at a given wavelength with a suitable liquid filter. These solutions are available as CRMs or can be prepared at the concentrations shown in Table 3 by using reagent-grade materials.
Table 3. Spectral Ranges of Selected Materials for Monitoring Stray Light
| Spectral Range (nm) |
Liquid or Solution |
|---|---|
| 190–205 |
Aqueous potassium chloride (12 g/L) |
| 210–259 |
Aqueous sodium iodide or potassium iodide (10 g/L) |
| 250–320 |
Acetone |
| 300–385 |
Aqueous sodium nitrite (50 g/L) |
When using a 5-mm path length cell (filled with the same filter) as the reference cell, and then measuring the 10-mm cell over the required spectral range, analysts can calculate the stray light value from the observed maximum absorbance using the formula:
S = 0.25 × 10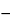 2A
2A
A
Acceptance criteria:
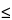 0.01. A
0.01. A 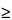 0.7A.
0.7A.
This procedure simply requires the 10-mm cell measurement to be referenced against the 5-mm cell (filled with the same filter) and therefore can be achieved by either chronological or spatial referencing in any type of spectrophotometer. Alternatively, analysts can measure the absorbance of the filters specified in Table 3 against the appropriate reference, and record the maximum absorbance value. (An S value of 0.01 is produced by an A value of 0.7A, which equates to a maximum absorbance value of 2A measured by this alternate procedure.) [Note—For some instruments where absorbance values greater than 3A cannot be reported directly, this procedure may require a two-step process whereby the sample beam initially is attenuated by a 1- to 2-A filter, the value of which is measured and recorded. After zeroing the instrument with this filter in place, measure the stray-light filter, and again record the absorbance value. The estimated stray light value is now the sum of these two absorbance readings.
Resolution
If accurate absorbance measurements must be made on benzenoid compounds or other compounds with sharp absorption bands (natural half-bandwidths of less than 15 nm), the spectral bandwidth of the spectrophotometer used should not be greater than 1/8th the natural half-bandwidth of the compound's absorption.
Determine the resolution of the spectrophotometer by using the following procedure. Measure the ratio of the absorbance of a 0.020% (v/v) solution of toluene in hexane (UV grade) at the maximum and minimum at about 269 and 266 nm, respectively, using hexane as the reference. The absorbance ratio obtained depends on the spectral bandwidth of the instrument. For most pharmacopeial quantitative purposes, a spectral bandwidth of 2 nm is sufficient, and the acceptance criteria for the ratio is NLT 1.3.
Table 4. Spectral Bandwidth and Measurement Temperature
| Temperature of Measurement |
Spectral Bandwidth |
||||
|---|---|---|---|---|---|
| 0.5 nm ± 0.1 nm |
1.0 nm ± 0.1 nm |
1.5 nm ± 0.1 nm |
2.0 nm ± 0.2 nm |
3.0 nm ± 0.2 nm |
|
| 20 ± 1 |
2.4–2.5 |
2.0–2.1 |
1.6–1.7 |
1.3–1.4 |
1.0–1.1 |
| 25 ± 1 |
2.3–2.4 |
1.9–2.0 |
1.6–1.7 |
1.3–1.4 |
1.0–1.1 |
| 30 ± 1 |
2.1–2.2 |
1.8–1.9 |
1.5–1.6 |
1.3–1.4 |
1.0–1.1 |
Suitable CRMs may also be used for this measurement. Alternatively, a suitable atomic line can be scanned in single-beam mode, and the peak width at half peak height can be determined. This peak width at half peak height equates to the bandwidth of the spectrophotometer.
Performance Qualification
The purpose of PQ is to determine that the instrument is capable of meeting the user's requirements for all the parameters that may affect the quality of the measurement and to ensure that it will function properly over extended periods of time.
PROCEDURE
With few exceptions, compendial spectrophotometric tests and assays call for comparison against a USP Reference Standard. This helps ensure measurement under identical conditions for the test specimen and the reference substance. These conditions could include wavelength setting, spectral bandwidth selection, cell placement and correction, and transmittance levels. Cells that exhibit identical transmittance at a given wavelength may differ considerably in transmittance at other wavelengths. Appropriate cell corrections should be established and used where required.
Comparisons of a test specimen with a reference standard are best made at a peak of spectral absorption for the compound concerned. Assays that prescribe spectrophotometry give the commonly accepted wavelength for peak spectral absorption of the substance in question. Different spectrophotometers may show minor variation in the apparent wavelength of this peak. Good practice demands that comparisons be made at the wavelength at which peak absorption occurs. Should this differ by more than ±1 nm (in the range 200–400 nm) or ±2 nm (in the range 400–800 nm) from the wavelength specified in the individual monograph, recalibration of the instrument may be indicated.
The expressions “similar preparation” and “similar solution” as used in tests and assays involving spectrophotometry indicate that the reference comparator, generally a USP Reference Standard, should be prepared and observed in an identical manner for all practical purposes to that used for the test specimen. Usually when analysts make up the solution of the specified reference standard, they prepare a solution of about (i.e., within 10%) the desired concentration, and they calculate the absorptivity on the basis of the exact amount weighed out. If a previously dried specimen of the reference standard has not been used, the absorptivity is calculated on the anhydrous basis. The expressions “concomitantly determine” and “concomitantly measure” as used in tests and assays involving spectrophotometry indicate that the absorbances of both the solution containing the test specimen and the solution containing the reference specimen, relative to the specified test blank, must be measured in immediate succession.
Sample Solution Preparation
For determinations using UV or visible spectrophotometry, the specimen generally is dissolved in a solvent. Unless otherwise directed in the monograph, analysts make determinations at room temperature using a path length of 1 cm. Many solvents are suitable for these ranges, including water, alcohols, lower hydrocarbons, ethers, and dilute solutions of strong acids and alkalis. Precautions should be taken to use solvents that are free from contaminants that absorb in the spectral region under examination. For the solvent, analysts typically should use water-free methanol or alcohol or alcohol denatured by the addition of methanol but without benzene or other interfering impurities. Solvents of special spectrophotometric quality, guaranteed to be free from contaminants, are available commercially from several sources. Some other analytical reagent-grade organic solvents may contain traces of impurities that absorb strongly in the UV region. New lots of these solvents should be checked for their transparency, and analysts should take care to use the same lot of solvent for preparation of the test solution, the standard solution, and the blank. The best practice is to use solvents that have NLT 40% transmittance (39.9%T = 0.399A) at the wavelength of interest.
Assays in the visible region usually call for concomitantly comparing the absorbance produced by the assay preparation with that produced by a standard preparation containing approximately an equal quantity of a USP Reference Standard. In some situations, analysts can omit the use of a reference standard (e.g., when spectrophotometric assays are made with routine frequency) when a suitable standard curve is available and is prepared with the appropriate USP Reference Standard, and when the substance assayed conforms to the Beer–Lambert law within the range of about 75%–125% of the final concentration used in the assay. Under these circumstances, the absorbance found in the assay may be interpolated on the standard curve, and the assay result can be calculated. Such standard curves should be confirmed frequently and always when a new spectrophotometer or new lots of reagents are put into use.
VALIDATION AND VERIFICATION
Validation
Validation is required when a UV-Vis method is intended for use as an alternative to the official procedure for testing an official article.
The objective of UV-Vis method validation is to demonstrate that the measurement is suitable for its intended purpose, including quantitative determination of the main component in a drug substance or a drug product (Category I assays), quantitative determination of impurities or limit tests (Category II), and identification tests (Category IV). Depending on the category of the test (see Table 2 in Validation of Compendial Procedures 1225), the analytical method validation process for UV-Vis requires testing for linearity, range, accuracy, specificity, precision, detection limit, quantitation limit, and robustness. These analytical performance characteristics apply to externally standardized procedures and those that use standard additions.
Chapter 1225 provides definitions and general guidance on analytical procedures validation without indicating specific validation criteria for each characteristic. The intention of the following sections is to provide the user with specific validation criteria that represent the minimum expectations for this technology. For each particular application, tighter criteria may be needed in order to demonstrate suitability for the intended use.
accuracy
For Category I, II, and III procedures, accuracy can be determined by conducting recovery studies with the appropriate matrix spiked with known concentrations of the analyte. Analysts also can compare assay results obtained using the UV-Vis procedure under validation to those from an established analytical procedure.
Validation criteria:
Precision
repeatability
The repeatability of the analytical procedure is assessed by measuring the concentrations of six independently prepared sample solutions at 100% of the assay test concentration. Alternatively, it can be assessed by measuring the concentrations of three replicates of three separate sample solutions at different concentrations. The three concentrations should be close enough so that the repeatability is constant across the concentration range. If this is done, the repeatability at the three concentrations is pooled for comparison to the acceptance criteria.
Validation criteria:
intermediate precision
The effect of random events on the analytical precision of the method must be established. Typical variables include performing the analysis on different days, using different instrumentation, and/or having the method performed by two or more analysts. At a minimum, any combination of at least two of these factors totaling six experiments will provide an estimation of intermediate precision.
Validation criteria:
specificity
In UV-Vis measurements, specificity is ensured by the use of a reference standard wherever possible and is demonstrated by the lack of interference from other components present in the matrix.
detection limit
The detection limit (DL) can be estimated by calculating the standard deviation of NLT 6 replicate measurements of a blank solution and multiplying by 3.3. Alternatively, the standard deviation can be determined from the error of the intercept from a calibration curve or by determining that the signal-to-noise ratio is >3.3. The estimated DL must be confirmed by analyzing samples at the calculated concentration.
quantitation limit
The quantitation limit (QL) can be estimated by calculating the standard deviation of NLT 6 replicate measurements of a blank solution and multiplying by 10. Alternatively, the standard deviation can be determined from the error of the intercept from a calibration curve or by determining that the signal-to-noise ratio is >10.
Measurement of a test solution prepared from a representative sample matrix spiked at the required QL concentration must be performed to confirm sufficient sensitivity and adequate precision. The observed signal-to-noise ratio at the required QL should be >10. [Note—A suitable procedure for measuring the signal-to-noise ratio is given in ASTM 1657-98 (2006) Standard Practice for the Testing of Variable-Wavelength Photometric Detectors Used in Liquid Chromatography.
Validation criteria:
linearity
A linear relationship between the analyte concentration and UV-Vis response must be demonstrated by preparation of NLT five standard solutions at concentrations encompassing the anticipated concentration of the test solution. The standard curve is then evaluated using appropriate statistical methods such as a least-squares regression. Deviation from linearity results from either instrumental or sample factors, or both, and can be reduced to acceptable levels by reducing the analyte concentration and thereby the associated absorbance values.
Validation criteria:
range
The operational range of an analytical instrument (and the analytical procedure as a whole) is the interval between the upper and lower concentrations (amounts) of analyte in the sample (including these concentrations) for which it has been demonstrated that the instrumental response function has a suitable level of precision, accuracy, and linearity.
Validation criteria:
robustness
The reliability of an analytical measurement is demonstrated by deliberate changes to experimental parameters. For UV-Vis this can include measuring the stability of the analyte under specified storage conditions, varying pH, and adding possible interfering species, to list a few examples. Robustness is determined concurrently using a suitable design for the experimental procedure.
indirect measurement requirements
For certain UV-Vis procedures, chromogenic reactions are employed. Generally the requirements for the analytical performance characteristics are used. In some instances, the required accuracy and precision criteria for the direct measurements may not be achievable. Under these circumstances, the accuracy and precision requirements can be widened by as much as 50%. Any such widening must be justified on scientific grounds and with documented evidence. It may be necessary to increase the amount of replication required to produce a scientifically sound reportable value.
Verification
Current US Good Manufacturing Practices regulations [21 CFR 211.194(a)(2)] indicate that users of analytical procedures described in USP–NF are not required to validate these procedures if provided in a monograph. Instead, they simply must verify their suitability under actual conditions of use.
The objective of a UV-Vis procedure verification is to demonstrate the suitability of a test procedure under actual conditions of use. Performance characteristics that verify the suitability of a UV-Vis procedure are similar to those required for any analytical procedure. A discussion of the applicable general principles is found in Verification of Compendial Procedures 1226. Verification is usually performed using a reference material and a well-defined matrix. Verification of compendial UV-Vis procedures includes at minimum the execution of the validation parameters for specificity, accuracy, precision, and quantitation limit, when appropriate, as indicated under Validation. USP38
USP38
1
The rounded values are taken from ASTM Standard E275-08.
2
NIST SRM 2034 is no longer available.
3
The rounded values are taken from the intrinsic wavelength standard absorption band data from Travis JC, Acosta JC, Andor G, et al. Intrinsic wavelength standard absorption bands in holmium oxide solution for UV/visible molecular absorption spectrophotometry. J Phys Chem Ref Data. 2005;34(1):41–57. The maximum 95% measurement uncertainty is ±0.06 nm.
4
ASTM E958 2011.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Margareth R.C. Marques, Ph.D.
Senior Scientific Liaison (301) 816-8106 |
(GCCA2010) General Chapters - Chemical Analysis |
USP38–NF33 Page 663
Pharmacopeial Forum: Volume No. 40(1)