



1. INTRODUCTION
Mass spectrometry (MS) is an integral part of modern pharmaceutical research and development in academic, industrial, and clinical laboratories. Trace analytical measurement (both qualitative and quantitative), specifically the demand for trace-mixture analysis, has increased the need for this powerful tool. In many cases, the analytical demands of trace-mixture sample analysis has made MS the method of choice for qualitative and quantitative assays for target analytes such as proteins, peptides, drug substances, metabolites, impurities, and degradation products. Because of its analytical capabilities, MS has found widespread application in the pharmaceutical industry. Specifically within the compendial context, MS has been, or has the potential to be, applied to both qualitative (identification tests) and quantitative measurements (assays).
For qualitative tests, MS-based methods can provide molecular mass information via the detection of the molecular ion or ions related to the molecular mass of the analyte as a first level or step of identification. A variety of instrumental methods permit molecular mass determinations for a wide range of materials, up to and including large biomolecules (e.g., proteins) and polymers. Along with the molecular mass information, MS also can provide unique structural information via the generation of fragment ions. The diversity of fragmentation approaches available in modern MS allows structurally significant fragmentation approaches for a wide variety of compendially relevant materials (e.g., small molecules and peptides).
Molecular mass thus can become a surrogate for confirmation or can even be used for the identification of a targeted compound, particularly when used in conjunction with an authentic standard or a chromatographic method. Advanced studies that involve one or two more dimensions of mass analysis also can be used to obtain specific structural detail (fragment ions that correspond to structurally unique portions of the target molecule) or more selectivity to enable powerful approaches for quantitation. Moreover, higher resolution methods that feature mass spectrometry and chromatography can routinely provide a benefit to the scientific community.
Modern analytical MS includes a diverse range of available instrumentation and experimental approaches. The specific mass spectrometer, and, of course, specific chemistries (i.e., sample preparation, chromatography, and ionization) define the final analytical procedure.
2. MASS SPECTROMETERS
The following sections describe the principles of MS, including the general layout of modern MS instrumentation used in the pharmaceutical industry and the specific components (sample introduction, ionization, and mass analyzer). This section also includes a brief description of the operational modes of a mass spectrometer, focusing on fragmentation production and analysis.
2.1 Overview
Analytical measurements that use MS generally involve the following processes: sample preparation, chromatography or sample introduction, ionization, and mass analysis and detection. The resulting output from the mass spectrometer is depicted in a mass spectrum, a survey of ions made in the ion source, and is represented in a graphic representation of mass-to-charge ratio (m/z) versus intensity.
Ions are separated by a property of mass such as m/z, where m is the mass of the ion and z is the number of charges on the ion. The method by which ions are separated typically defines the mass spectrometer type. Regardless of type, the mass analyzer separates ions according to the m/z. The mass analyzer continuously acquires data across a predefined range of masses to generate the resulting mass spectra.
2.2 General Layout
A general layout of a mass spectrometer platform for pharmaceutical analysis is shown in Figure 1. Samples are prepared via a procedure defined for the specific sample and analyte. The resulting sample then is injected into the mass spectrometer via a chromatographic system such as high-performance liquid chromatography (HPLC) or gas chromatography (GC) or via direct injection. From the inlet, the discrete analytes are ionized for subsequent analysis by the mass spectrometer. All mass spectrometers require four components:
- Sample introduction technique
- An ionization source to charge the analyte
- A mass analyzer to separate the analytes on a mass/charge (m/z) scale
- A detector to measure the ions.
+'&img=../../images/v38332/c1736-fig1_2s37.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 1. General MS system. HPLC = high-performance liquid chromatography; UV = ultraviolet detector. (Courtesy of Milestone Development Services, Newtown, PA)
MS can be categorized by the dimensions of mass analysis: single-stage mass spectrometers, hybrid or tandem mass spectrometers (MS/MS), and multiple stages of mass analysis (MSn). These mass spectrometer formats are described in this section.
2.2.1 single-stage ms
A single-stage mass spectrometer provides one dimension of mass analysis. As shown in the schematic in Figure 2, a single mass spectrometer can be viewed simply as providing a survey of all ions generated in the ion source. For example, if a 10-component mixture is injected into a chromatograph and each component is separated with adequate resolution, then the mass spectrometer would provide discrete measurement of mass-to-charge of each of the 10 components. The resulting mass spectrum of each component is generated for subsequent interpretation and assignment of molecular mass.
+'&img=../../images/v38332/c1736-fig2_2s37.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 2. One-dimensional MS scan. Q1 = single-stage mass spectrometer or a single stage of analysis. (Courtesy of Milestone Development Services, Newtown, PA)
2.2.2 tandem mass spectrometry (ms/ms)
A system capable of carrying out two sequential m/z analysis events (i.e., a tandem mass spectrometer) provides increased selectivity for detailed structure elucidation or quantitative analysis.
2.2.3 multiple stages (msn)
A unique feature of some MS formats is the capability to perform multiple stages of MS and to generate further fragment information. These mass spectrometers are capable of isolating an ion of interest, inducing fragmentation, isolating a specific product ion and then repeating this process on the resulting or selected product ion.
2.3 Sample Introduction
Several approaches can be used to introduce the sample into the mass spectrometer. The direct, infusion, and chromatographic procedures are described in this section.
2.3.1 direct introduction
For some sample types and applications, a direct insertion probe is used to introduce the sample into the mass spectrometer. The sample, usually a pure compound or relatively pure compound, is dissolved in an appropriate solvent. A small amount of the sample (typically 1 µL or less) is deposited onto the probe. Typically, the probe consists of a metal filament, such as platinum, located at its tip. The metal filament is heated, and the sample is desorbed into the ion source of the mass spectrometer. The probe then is removed from the mass spectrometer and is prepared for the next sample.
2.3.2 infusion introduction
Sample introduction via infusion typically is done to provide a relatively long analysis time or perhaps to conduct a quick survey of a sample. Infusion introduction also can be done when analysts optimize the instrumental conditions (source and mass spectrometer operational parameters) for a specific analyte as well as to obtain greater numbers of spectra. This sample introduction method may require more sample than conventional flow rates. However, infusion sample introduction is widely practiced for nanospray ionization applications for the analysis of proteins and peptides as well as during specialized nanospray applications with small molecules in drug metabolism to determine structure or equimolar response ratios.
2.3.3 chromatographic introduction
The use of chromatography methods for sample introduction into the mass spectrometer is perhaps the most common approach.
2.3.3.1 Gas chromatography:
GC procedures are preferred for nonpolar, volatile analytes (see Chromatography  621
621 . Samples contained in solvent are injected onto the injection port of the GC. The sample is volatilized, and a nonreactive, inert gas such as helium carries the sample through the GC column, which is contained in a temperature-controlled oven. The combination of carrier gas and heat moves the analyte through the column. Capillary GC columns are commonly used in pharmaceutical applications. Molecules separate within the capillary column and are introduced directly into the ion source of the mass spectrometer.
. Samples contained in solvent are injected onto the injection port of the GC. The sample is volatilized, and a nonreactive, inert gas such as helium carries the sample through the GC column, which is contained in a temperature-controlled oven. The combination of carrier gas and heat moves the analyte through the column. Capillary GC columns are commonly used in pharmaceutical applications. Molecules separate within the capillary column and are introduced directly into the ion source of the mass spectrometer.
2.3.3.2 High-performance liquid chromatography:
A popular approach for sample introduction into the mass spectrometer involves HPLC (see 621). HPLC procedures are preferred for nonvolatile and thermally labile analytes, but are suitable for use with any analyte that is readily ionizable in a solution environment with the appropriate chemical modifiers. Samples are prepared in solution and then injected onto the HPLC column. Analytes are separated based on the partitioning between the mobile phase and stationary phase and are introduced into the ion source of the mass spectrometer. The effluent is volatilized in the ion source and ionization occurs. The subsequent ions then are introduced into the mass spectrometer for analysis.
2.3.3.3 Capillary electrophoresis:
Capillary electrophoresis (CE), also called capillary zone electrophoresis (CZE), is a separation method that exploits subtle differences in the ionic composition of analytes to separate them based on electrophoretic mobility in a conductive liquid. Details regarding the principles and use of this methodology can be found in Capillary Electrophoresis 1053. During the past 15 years this separation method periodically has been used with MS as an alternative to LC–MS for certain classes of compounds. However, with the widespread use of HPLC in analytical and MS laboratories, CE-coupled MS methods often are superseded by LC–MS methods.
2.4 Ion Polarity
Ionization involves the process of converting the analyte into the gas phase and depositing a charge onto the molecule. The final charge of the analyte determines the ion polarity to be used in the mass analysis. Generally, selection of the ionization mode depends on the ability of the desired analyte to accept or lose a proton during the ionization process. MS can be performed in the positive or negative ion polarity mode.
2.4.1 positive ion mode
Many MS pharmaceutical analyses use the positive ion mode. For example, compounds that contain a basic functional group such as an amine (e.g., proteins and peptides) are excellent candidates for MS analyses in the positive ion mode because under acidic conditions these compounds readily accept a proton to form positively charged ions.
2.4.2 negative ion mode
The negative ion mode is well suited for compounds that can easily lose a proton. Compounds that contain a carboxylic acid, phosphate, or a sugar, for example, are good candidates for negative ion MS.
2.5 Ionization Procedures
A variety of ionization procedures can be used with a mass spectrometer for pharmaceutical analysis. The various ionization procedures are described in this section.
2.5.1 electron ionization
Electron ionization (EI) is used mostly with GC applications when the analytes of interest are nonpolar and are easily volatilized. Mass spectra produced by EI procedures are characterized by extensive fragmentation. Because EI typically produces extensive fragmentation, EI is considered a hard-ionization mode. Mass spectra are obtained when a 70 eV electron beam enters the source and impacts the analyte molecules present in the gas phase:
M + e ® M+· + 2e
® M+· + 2e
Nearly all EI applications are conducted in the positive ion mode. These spectra are highly reproducible. Thus, EI spectral libraries can be used to determine the structure and to confirm the identity of unknown compounds. Additionally, fragmentation patterns have been extensively studied for electron ionization and can help determine structure of unknown compounds.
2.5.2 chemical ionization
Chemical ionization (CI) procedures rely on electron ionization of reagent ions such as methane, ammonia, or isobutene. The reagent ions react with the analyte molecules (ion–molecule reaction) in the source of the mass spectrometer. In the positive ion mode, analyte molecules are ionized via proton transfer and/or adduct formation, producing even electron molecular ion species or adduct ions. Depending on the analyte, both positive and negative ion spectra can be obtained with CI procedures. CI is considered a softer ionization (less fragmentation of the molecular ions) mode than EI although some approaches can induce fragmentation depending on the CI gas chosen and the structure of the analyte molecule. CI is very useful for reactive and unstable compounds where a molecular mass determination is desired. Representative CI ionization reactions for both GC–MS and LC–MS methods follow.
Protonated molecule formation (GC–MS example):
CH4 + e ® CH4+ + 2e
CH4+ + CH4 ® CH5+ + CH3
CH3+ + CH4 ® C2H5+ + H2
M + C2H5+ ® [M + H]+ + C2H4
Protonated molecule formation (LC–MS example):
M + NH4+ ® NH3 + [M + H]+
Adduct ion formation (LC–MS example):
M + NH4+ ® [M + NH4]+
2.5.3 atmospheric pressure ionization
Atmospheric pressure ionization (API) procedures allow the direct introduction of samples into the mass spectrometer from a liquid interface such as an HPLC. The two most common forms of API used in pharmaceutical analyses are electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI).
2.5.3.1 Electrospray ionization:
ESI is a method that produces fine charged droplets of a liquid phase that carries the analyte of interest. The liquid phase typically is a volatile combination of water and organic solvent (e.g., acetonitrile or methanol). A small percentage of a reagent (e.g., 0.1% formic acid) also is included to increase the conductivity of the solution. ESI typically is used with HPLC and involves the nebulization of the sample delivered at flow rates that range from nL/min to mL/min to produce a fine spray of droplets (radius = 0.5–1.0 µm). Solvent evaporation results in an increased charge concentration at the droplet surface until ions are liberated directly from the droplet. Ions are transported or focused directly into the mass spectrometer, and the resulting spectra contain ions that are typically indicative of the molecular mass of the analyte.
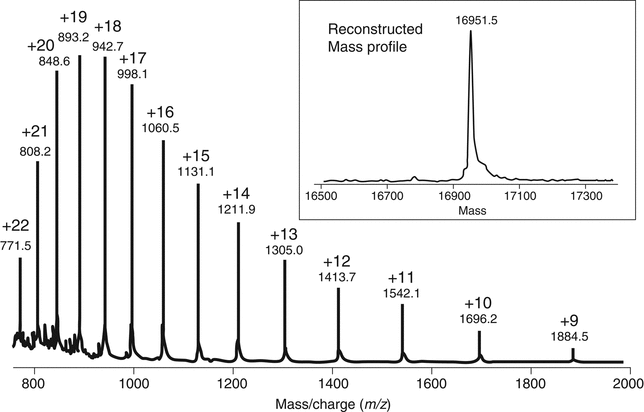
ESI often is referred to as a soft-ionization procedure because typically it does not result in fragmentation of the analyte during the ionization process. The development of this ionization technology was crucial for the analysis of large biopolymers (e.g., proteins) because it allows the addition of multiple charges to a single molecule, thus bringing the m/z of the analyte into a suitable m/z range for many types of mass analyzers. For example, Figure 3 shows the ESI charge state distribution for equine heart apomyoglobin as analyzed on a simple single quadrupole instrument with an upper scan limit of m/z 2000. The series of multiply charged ions allows the spectrum to be collected within the instrument's working m/z range. The ability to calculate the intact mass of the protein from a combination of all the charge states allows a more accurate average mass (see inset in Figure 3) to be determined (within 0.1% on a simple quadrupole instrument). Thus ESI methods are critically important for the analysis of large biomolecules.
+'&img=../../images/v38332/c1736-fig3.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 3. Electrospray ionization mass spectrum for equine heart apomyoglobin.
2.5.3.2 Atmospheric pressure chemical ionization:
APCI procedures are used with HPLC and involve the nebulization and heating of the sample to liberate neutral molecules. A corona discharge produces reagent ions (e.g., H3O+ or NH4+) from the mobile phase. The reagent ions react with analyte molecules via proton transfer or adduct formation. The resulting positive ion spectrum typically contains [M + H]+ ions that are indicative of molecular mass and fragment ions that correspond to unique substructure(s) of the analyte molecule. Negative ion spectra are generated when reagent ions (e.g., OH or CH3COO) react with the analyte molecule to produce [M H] ions.
2.5.4 matrix-assisted laser desorption ionization
Matrix-assisted laser desorption ionization (MALDI) is a soft-ionization procedure used primarily for biomolecules that relies on the addition of a chemical matrix dried with the analyte of interest. This matrix compound can absorb laser energy at a particular wavelength during the laser ablation process. By a mechanism that has not been fully elucidated, some of the ions generated in the matrix can transfer protons to the analyte, and the resulting gas-phase ions are focused into the mass spectrometer. Specific matrices have been empirically developed for various classes of analytes, including 3,5-dimethoxy-4-hydroxycinnamic acid (sinapinic acid) and 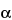 -cyano-4-hydroxycinnamic acid for proteins and peptides and picolinic acid for oligonucleotides. Unlike ESI, MALDI typically produces lower charge-state ions, with singly charged ions being the most favorable.
-cyano-4-hydroxycinnamic acid for proteins and peptides and picolinic acid for oligonucleotides. Unlike ESI, MALDI typically produces lower charge-state ions, with singly charged ions being the most favorable.
2.5.5 ambient ionization procedures
Ambient ionization generally refers to a collection of MS procedures that permit direct sampling and interrogation of analytes from sample matrices or surfaces under ambient conditions with little or no pretreatment. In recent years, the number and type of ambient ionization procedures have rapidly expanded. This is evident from the proliferation in recent literature of acronyms that represent various ambient ionization procedures. Ambient ionization has enjoyed broad application in fields that include forensics; food safety; monitoring of environmental contaminants; polymers; fuels; detection of explosives and drugs of abuse; molecular imaging of surfaces and tissues; profiling and characterization of metabolites, proteins, and biomolecules; as well as monitoring of chemical reactions and processes.
Ambient ionization procedures typically produce analyte ions directly from sample surfaces or require initial production of analytes that subsequently are ionized by any of several processes. Therefore, ambient ionization procedures can be broadly categorized as representing either direct or multistage ionization mechanisms. Direct ionization procedures generate analyte ions from a sample solution or droplet in an electric field, or desorb analyte ions by impinging a sample surface with charged droplets or solvent ions, photons, or metastable atoms. Multistage ionization procedures initially produce analyte particle or droplets with a liquid or gas stream, thermal desorption, irradiation or ablation with a laser, or nebulization. Analytes subsequently react with a charged species or with metastable atoms generated by ESI, APCI, or photoionization to produce analyte ions.
Table 1 is a representative, but not comprehensive, listing of ambient ionization procedures and illustrates the diversity and similarity among various approaches.
Table 1. Ambient Ionization Procedures
| Acronym | Description | Mechanisms |
|---|---|---|
| APGDDI | Atmospheric pressure glow discharge desorption ionization |
Thermal desorption, gas discharge ionization |
| APPeIa | Atmospheric pressure Penning ionization | Similar to APCI |
| AP–TD/ESIa | Atmospheric pressure thermal desorption/electrospray ionization | Thermal desorption, ESI |
| ASAPa | Atmospheric pressure solids analysis probe | Thermal desorption |
| DAPCI | Desorption atmospheric pressure chemical ionization |
Thermal desorption, APCI |
| DAPPIa | Desorption atmospheric pressure photoionization | Thermal desorption, photoionization |
| DARTa | Direct analysis in real time | Gas discharge ionization |
| DBDI | Dielectric barrier discharge ionization |
Thermal desorption, gas discharge ionization |
| DCBI | Desorption corona beam ionization | Thermal desorption, APCI |
| DESI | Desorption electrospray ionization | Similar to ESI |
| EADESI | Electrode-assisted desorption electrospray ionization |
Similar to ESI |
| EASI | Easy ambient sonic spray ionization | Supersonic spray ionization |
| EESIa | Extractive electrospray ionization | Similar to ESI |
| ELDIa | Electrospray laser desorption ionization |
Laser desorption or ablation, ESI |
| FD–ESIa | Fused droplet electrospray ionization |
Similar to ESI |
| IR–LADESIa | Infrared laser-assisted desorption electrospray ionization |
Laser desorption or ablation, ESI |
| LAESIa | Laser ablation electrospray ionization |
Laser desorption or ablation, ESI |
| LD–APCIa | Laser desorption atmospheric pressure chemical ionization | Laser desorption, APCI |
| LD–ESIa | Laser desorption electrospray ionization | Laser desorption, ESI |
| LDSPIa | Laser desorption spray postionization | Laser desorption or ablation |
| LDTDa | Laser diode thermal desorption |
Thermal desorption |
| LEMSa | Laser electrospray mass spectrometry |
Laser desorption or ablation |
| LESAa | Liquid extraction surface analysis | Similar to ESI, surface extraction with solvent droplet |
| LIAD–ESIa | Laser-induced acoustic desorption electrospray ionization | Laser desorption or ablation, similar to ESI |
| LPI–MSa | Liquid surface Penning ionization mass spectrometry |
Similar to APCI |
| LSI | Laser spray ionization | Laser desorption or ablation |
| MALDESIa | Matrix-assisted laser desorption electrospray ionization |
Laser desorption or ablation |
| PADI | Plasma-assisted desorption ionization |
Thermal desorption, gas discharge ionization |
| SESIa | Secondary electrospray ionization | Similar to ESI |
| TDAMSa | Thermal desorption based ambient mass spectrometry | Thermal desorption |
| TD/APCIa | Thermal desorption/ atmospheric pressure chemical ionization |
Thermal desorption, similar to APCI |
|
a
Multistage ionization procedure.
|
||
3. MASS ANALYSIS
The method by which ions are separated typically defines the mass spectrometer type. Essentially, the mass analyzer is responsible for filtering the ions generated during the ionization process. The various mass analyzers that typically are used in the pharmaceutical industry are described in this section.
3.1 Quadrupole
A quadrupole mass spectrometer consists of a set of four parallel rods. When a combination of constant (DC) and alternating (AC) voltage are applied to the opposing rods respectively, the resulting electric fields allow ions of a specific m/z to stably transit the quadruple and to pass through to the detector. Quadrupole mass spectrometers are relatively low-cost instruments and provide good qualitative and quantitative analytical capabilities. Generally, quadrupole mass spectrometers are limited to production of low-resolution mass spectra.
3.2 Magnetic Sector
A magnetic sector mass spectrometer filters ions by the means of the application of a magnetic field. The magnetic field is varied, and ions are deflected to follow a curved path so that ions with different m/z ratios are separated.
3.3 Ion Traps and Ion Cyclotron Resonance
Most commercial mass spectrometers fall broadly into two categories: those that rely on continuous beams of ions being sent to a detector for sequential m/z detection (like cars on a highway driving from point A to point B), and those devices that “trap” ions in discrete, repeating orbits (like cars on a race track). The latter devices are broadly referred to as ion traps. The orbital paths are complex and unique to each device. The ions can be trapped by static electric fields (DC voltage), dynamic quadrupole electric fields [radio frequency (RF) AC voltage], magnetic fields, or some combination of the types. Detection of the ions may take place by sequential ion ejection (after trapping, by m/z) or by detection of all ions simultaneously [while trapped, i.e., by image current detection and subsequent Fourier transformation (FT) of the data]. Examples include the popular linear and 3D ion trap MS systems (RF AC voltage only with ion ejection for detection), the FT Orbital Trapping MS (which uses DC voltage and a quadrupole field for trapping, with image current detection), and the less common FT Ion Cyclotron Resonance MS (which uses a superconducting magnet to provide the primary trapping field). The FT devices generally provide very high mass resolution (e.g., 105–106 at m/z 400) at reasonable scan speeds (1 Hz), and the RF-based devices generally offer fast scan rates (0.1 Hz) but lower resolution (103). Lastly, the most popular FT equipment is now hybridized with a quadrupole RF device (especially the linear trap) to yield instrumentation with multiplexed experiment capabilities (multiple sequential MS–MS experiments with accurate m/z determinations for structure elucidation).
3.3.1 time-of-flight
A time-of-flight (TOF) mass spectrometer uses differences in transit time through a field-free drift region to separate ions. Ions generated in the ion source are pulsed into the field-free drift region (flight tube) by an electric field. Lighter ions have a higher velocity and reach the detector sooner. The TOF mass spectrometer has benefited significantly from the use of fast electronics and fast computers to provide systems characterized by high speed, high sensitivity, and high resolution.
4. MS/MS AND MSn SPECTROMETRY INSTRUMENTATION
A wide variety of tandem MS instruments are available to the pharmaceutical research and development community. Each of the tandem instruments has some unique features that make it particularly suited for some specific applications, but this does not preclude the use of other tandem instruments to meet a specific analytical need. The appropriate selection of a tandem instrument that is suitable to meet a specific need depends on the type of analysis required (e.g., quantitation or structural elucidation), a variety of sample-specific factors (e.g., analyte concentration or the complexity of the matrix), and whether sample fractionation is incorporated before the introduction in the mass spectrometer (e.g., by LC–MS). A brief summary of some of the common types of tandem instruments follows, highlighting some of the typical applications.
4.1 MS/MS and MSn Spectrometers
4.1.1 triple quadrupole
The triple quadrupole mass spectrometer is used for both qualitative and quantitative analysis. A triple quadrupole mass spectrometer features a first stage of mass analysis (Q1) for the selection of a precursor ion, followed by an RF-only collision quadrupole region (Q2). A second stage of mass analysis (Q3) is used for product ion analysis. Most quantitative LC–MS-based analyses feature the use of triple quadrupole mass spectrometers. Furthermore, the triple quadrupole mass spectrometer is capable of providing all the most common qualitative scan modes: full scan, product ion, precursor ion, and neutral loss (see Figure 4 and 4.2 MS/MS and MSn Spectrometry Operational Modes).
+'&img=../../images/v38332/c1736-fig4a_2s37.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
A. Full Scan
+'&img=../../images/v38332/c1736-fig4b_2s37.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
B. Product Ion Scan
+'&img=../../images/v38332/c1736-fig4c_2s37.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
C. Precursor Ion Scan
+'&img=../../images/v38332/c1736-fig4d_2s37.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
D. Neutral Loss ScanFigure 4. Triple quadrupole MS. (A) Full scan—Q1 scans the ions produced in the ion source. This scan also can be conducted with Q3. (B) Product ion scan—Q1 is set to select a specific molecular ion produced in the ion source. Fragmentation of the ion(s) selected in Q1 occurs in the collision cell (Q2). Q3 scans the resulting fragments. The product ion spectrum contains all the fragments of the selected precursor ion(s). (C) Precursor ion scan—Q1 scans the ions produced in the ion source. Ions fragment in the collision cell (Q2). Q3 is set to select a specific product ion (derived from the product ion spectrum). The scan results in a spectrum that contains all precursor ions that generate the specific product ion. (D) Neutral loss scan—Q1 scans the ions produced in the ion source, and ions fragment in the collision cell (Q2). Q3 also scans, but at a difference in mass equal to a selected neutral loss (as derived from the product ion spectrum). The scan results in a spectrum that contains all molecular ions that generate a specific neutral loss fragment. (Courtesy of Milestone Development Services, Newtown, PA)
4.1.2 tof–tof system
A tandem configuration for TOF systems typically is used for the characterization and sequencing of peptides and other biomolecules from fairly simple mixtures (e.g., proteolytic digestion of a single-protein therapeutic). These systems often are configured for ionization by MALDI, which allows peptide sequence confirmation without the need for an initial separation. The relative ease of use of such instruments and the robust fragmentation data collected for confirmation of peptides may allow TOF–TOF instruments to be used for monograph identification testing in the future.
4.1.3 magnetic sector
Tandem configurations for magnetic sector instruments traditionally have been used for analyses that require high resolution and mass accuracy, such as structural elucidation of unknowns. However, because of greater resolution and mass accuracy becoming available in other tandem MS platforms (e.g., ion traps and quad-TOF devices), magnetic sector instruments are now less commonly used for these purposes.
4.1.4 ion trap
The tandem configuration for ion trap systems allows the isolation and fragmentation of ions in a time-dependent manner, unlike other tandem instruments that have spatially distinct mass analyzers in the path of the ion beam. One advantage of the trap-isolate-fragment-detect mechanism in an ion-trap instrument is the ability to re-isolate product ions from the initial precursor ions for further fragmentation. This sequential fragmentation allows more detailed structural determination to assist in elucidating structural unknowns. For protein and peptide applications, ion-trap instruments have been widely used to identify and sequence peptides and to characterize protein modifications (e.g., phosphorylation and glycosylation).
4.1.5 fourier-transform ion cyclotron resonance
Fourier-transform ion cyclotron resonance (FT–ICR) mass spectrometers, like ion traps, facilitate a time-dependent isolation of ions. FT–ICR mass analyzers offer high resolution (typically 105106) and can achieve sub-ppm mass accuracy, which permits determination of accurate mass and elemental composition for unknown compounds with a high degree of certainty. Ions can be selectively excited or ejected from the ICR cell by generation of a waveform that contains resonant frequencies of the m/z values of the ions of interest. Several fragmentation procedures are available for achieving the structural characterization of small and large molecules with FT–ICR MS, including collision-induced dissociation (CID), electron capture dissociation (ECD), and infrared multiphoton dissociation (IRMPD). Some commercial FT–ICR MS systems use a tandem ion trap to isolate and fragment precursor ions, the product ions of which are subsequently transferred to the ICR cell for detection or additional fragmentation by other procedures. The high resolution and mass accuracy of FT–ICR MS make this instrument particularly well suited for the structural characterization of components that comprise highly complex mixtures of small and large organic molecules. FT–ICR MS also has been widely used in the characterization of lipids and carbohydrates and in the characterization and sequencing of peptides and proteins, including examination of unique features such as post-translational modifications.
4.1.6 hybrids
A variety of hybrid tandem mass spectrometers have emerged to either enhance specific applications or to broaden the overall utility of the instrument in a variety of applications. Currently the most common hybrid instrument used for pharmaceutical characterization is the quadrupole TOF (Quad-TOF) MS, which combines the scanning speed of a quadrupole mass filter with the resolving power and mass accuracy of the TOF mass analyzer. This combination is well suited for structural elucidation because the improved resolution and mass accuracy of the TOF (by comparison with a quadrupole) allow a greater degree of certainty in determining the probable elemental makeup of both the precursor and product ions. In addition, the use of a quadrupole mass filter in this tandem configuration allows scanning functions like those outlined for a triple-quadrupole instrument (Figure 4), although additional postprocessing of the data is required. An increasing number of hybrid configurations have been marketed to take advantage of the unique feature of the combinations for resolution, mass accuracy, and quantitation. Some of these configurations include Quadrupole–Trap, Trap–TOF, Trap–IRC, and orbital trapping mass spectrometers.
4.2 MS/MS and MSn Spectrometry Operational Modes
4.2.1 fragmentation production modes
Collision-Induced Dissociation (CID) or Collisionally Activated Dissociation (CAD) is the process by which a selected precursor ion undergoes collisions with neutral gas molecules in a collision region to yield product ions. Fragmentation occurs at relatively low energies (1–100 eV) primarily at the site(s) of ionization to yield a fragment ion and a neutral molecule. Nitrogen, argon, or helium typically is used as the collision gas with collision energies in the 10–50 eV range.
Electron Capture Dissociation (ECD) and Electron Transfer Dissociation (ETD) are two distinct but related fragmentation methods used primarily to fragment proteins and peptides to generate c- and z-type ions that are useful for determining protein sequence and characterization of protein modifications. The fragmentation in each case relies on the introduction of either a free electron (ECD) or a radical anion (ETD) into the gas phase of positively charged molecules to induce chemical fragmentation. The advantage of these methods for proteins and peptides versus CID is that the chemical fragmentation occurs almost exclusively along the peptide backbone, thereby preserving the position and context of protein modification that often are lost during CID.
4.2.2 fragmentation analysis modes
The most common MS/MS scan modes are described in this section.
4.2.2.1 Product ion:
The product ion scan mode is illustrated in Figure 4B for a triple-quadropole (QQQ) instrument. Application of the product ion scan mode is performed when the first mass analyzer (Q1) is held at a specific m/z. This ion, typically the protonated molecule, [M + H]+, is fragmented in the collision cell (Q2). The second mass analyzer (Q3) scans the resulting product ions. For a trapping device such as an ion trap mass spectrometer, ions of a specific m/z are isolated and are collisionally activated, followed by scanning of the product ions. In either case, the resulting product ions' MS–MS spectrum contains the diagnostic fragment ions indicative of the characteristic substructures of the selected analyte. This scan mode requires that a full scan mass spectrum be obtained before the product ion experiment to determine the appropriate selection of the molecular ion (typically [M + H]+). The product ion scan mode is available in all mass spectrometers capable of MS–MS operation.
4.2.2.2 Precursor ion:
The precursor ion scan mode is illustrated in Figure 4C. Application of the precursor ion scan mode is performed when the first mass analyzer (Q1) is scanned and allows all ions created in the ion source to pass into the collision cell (Q2). The second mass analyzer (Q3) is held at a specific m/z ratio that corresponds to a diagnostic or unique fragment ion of the molecule or class of molecules. The resulting precursor ion MS–MS spectrum contains all molecular ions that contain a diagnostic fragment ion. The precursor ion scan mode requires that the product ion experiment be performed to confirm the diagnostic fragment ion(s). This scan mode can be used to screen for molecules that contain a specific substructural feature such as a peptide residue. The precursor ion scan mode is available on triple-quadrupole, magnetic sector, and some hybrid instrumentation.
4.2.2.3 Neutral loss:
The neutral loss scan mode is illustrated in Figure 4D. Application of the neutral loss scan mode is performed when both mass analyzers (Q1 and Q3) are scanned at the same rate. The second mass analyzer (Q3) is offset (lower) from the first by a constant m/z difference. Molecular ions enter Q1 and fragment in the collision cell (Q2), and the resulting fragment ions are detected after passage through Q3. Because of the offset scan function described above, the resulting spectrum presents all molecular ions that have undergone the selected neutral loss. This scan mode also requires that the product ion scan be performed to confirm the diagnostic neutral loss(es). Like the precursor ion scan mode, this scan mode is used to screen for molecules with a diagnostic structural feature such as phosphate or glucuronide conjugation. The neutral loss ion scan mode is available on triple-quadrupole, magnetic sector, and some hybrid instrumentation.
4.2.2.4 MSn:
The MSn acquisition mode is essentially a sequential application of the product ion scan discussed in 4.2.2.1 Product ion. That is, it is used to further fragment product ions from a previous product ion experiment. Thus it can be utilized to obtain additional structural information for identification of analytes. This acquisition mode typically is available only on ion trap or FT–ICR instruments.
5. QUALITATIVE ANALYSIS
MS is a powerful method and important tool for structure identification because of its ability to provide information about the mass, elemental composition, and structural features of known and unknown molecular entities. In its simplest terms, an MS separates and mass-measures ions related to the sample. A mass spectrometer separates, detects, and records the presence of ions, not neutral molecules. For the purposes of small-molecule organic MS, the charge state (z) typically is 1.
A wide range of mass spectrometers and ionization interfaces are available for specific types of MS measurements. Thus a diverse range of experimental approaches can be employed for any given MS measurement needed. Some of the more common and applicable approaches are presented in this section. The appropriateness of a given MS experimental approach is demonstrated by the respective validation data provided in the submission supporting a new monograph or monograph procedure.
5.1 The Mass Spectrum
The mass spectrum typically is displayed as a plot of m/z on the abscissa versus ion intensity as the ordinate and frequently is normalized to the most intense ion in the spectrum. Some characteristic features in a typical full-scan mass spectrum include (depending on ionization procedure) the molecular ion (e.g., M+), protonated or deprotonated molecules ([M + H]+ or [M H]), or adduct ions (e.g., [M + NH4]+), all of which are indicative of the molecular mass of the analyte. Mass spectra may be more or less complex, depending on the specific analyte(s) and ions detected. EI mass spectra typically are rich in fragment ions, but API and soft-ionization procedures may produce spectra that are sparse in fragment ions but often feature more prominent or base peak ions that are indicative of MW. Therefore, the interpretation of mass spectra may be more or less straightforward. Typically, the availability of any supporting information about the sample (e.g., origin, history, preparation, stability, solubility) can greatly assist with interpretation. Two primary considerations merit initial discussion because of their importance in the interpretation of mass spectra: resolution and mass accuracy.
5.1.1 mass resolution
In MS, resolution is the extent to which two ions of adjacent m/z can be distinguished from each other, and has been expressed in different ways. Two adjacent peaks in a spectrum at values m and m Dm may be separated by a valley, which at its lowest point is a specified maximum amount of overlap between the peaks. The resolution would be expressed as m/Dm. For example, the term “10% valley” describes a 10% amount of overlap between two peaks and is appropriate when the peaks are of equal height and shape. In practice, however, adjacent peaks of equal height and shape are rarely observed. Therefore, resolution also can be calculated by using the apparent width of a single peak at a given point in mass range as Dm, and it often is expressed as the measured width of a peak at half-height (full-width half-height maximum, or FWHM). Resolution is an important factor in establishing mass accuracy, evaluating isotope patterns and abundance, determining charge state of multiply charged ions, and distinguishing nominally isobaric interferences.
Nominal mass spectrometers instruments typically provide mass resolution on the order of 1000, whereas for high-resolution mass spectrometry (HRMS) this can range from 5000 to 1,000,000. Improved resolution includes the ability to separate and distinguish isobaric interferences (analytes with the same nominal mass but with different empirical formulae) and to mine complex data sets more efficiently (using increased specificity).
5.1.1.1 Nominal resolution:
A number of different types of mass spectrometers are capable of generating nominal resolution mass spectra. Probably the most common mass spectrometer employed for nominal mass resolution work is the quadrupole mass spectrometer. In a quadrupole mass spectrometer, ions pass through a set of four rods such that opposing rods ramp through RF or DC, effectively scanning through the desired m/z range and allowing only a single m/z to pass at any given instant. The net effect is to obtain nominal mass resolution (e.g., m/z 400 can be reliably distinguished from m/z 399 and m/z 401) and to obtain a spectrum that can define monoisotopic mass and associated isotopic cluster (e.g., A + 1 and A + 2). With this type of mass spectrometer (nominal resolution), the primary information obtained is an indication of the molecular mass of the analyte and, depending on the experimental approach used (e.g., EI ionization), ion fragments related to the structure of the analyte.
5.1.1.2 High resolution:
HRMS has important advantages for the identification of analytes. The various forms of this technology allow determination of the mass of small molecules to the third or fourth decimal place (i.e., approximately 0.1 to 10 ppm mass error). This provides an added degree of specificity (certainty) that, when combined with isotope ratio information and chemical sense, results in elucidation of the analyte's empirical formula as a starting point for further analysis. Thus HRMS offers advantages compared to nominal MS, for which nominal mass is the starting point. HRMS can be performed with a number of different technologies, including TOF–MS, as well as orbital-trapping MS and ion cyclotron resonance MS, where the latter two approaches utilize Fourier transformation to process the raw data. These systems have different advantages, including improved sensitivity for full spectrum data collection (versus scanning instruments like quadrupoles), as well as resolution, mass accuracy, and calculation of the isotopic ratio intensities.
5.1.2 mass accuracy
Mass accuracy is the comparison of an experimentally determined m/z to the true m/z ratio. Mass accuracy is influenced by a number of factors, including mass resolution, signal abundance, calibration type and algorithms, choice and number of calibrants used, interferences such as chemical noise, and linear dynamic range inherent with a particular type of mass spectrometer. Mass accuracy may be expressed as millimass unit (0.001 µ) differential or ppm. Mass accuracy, as ppm, is given by the following:
Dmacc = mtrue mmeas
ppm = (Dmacc/mtrue) × 106
mmeas = measured value
mtrue = calculated or true mass
Although resolution and accuracy may not be directly correlated, HRMS typically allows improved assignment of exact mass. In practice, high resolution and high mass accuracy are both necessary to minimize error introduced into the mass measurement by isobaric interferences. Sufficiently high resolution and mass accuracy together can facilitate assignment of elemental composition to ions in a mass spectrum, assisting with confirmation or elucidation of structure, respectively, for known or unknown compounds.
5.2 Interpretation of Mass Spectra
When interpreting the mass spectra of unknown compounds, analysts typically can differentiate unknowns into two broad categories.
The first category, representing the situation more commonly encountered, can be described as known unknowns or compounds for which supportive information may be available or can be determined by comparison with standards, related substances, metabolites, synthetic impurities, or intermediates. In this case, the objective is verification of a compound's identity by demonstrating its consistency with a standard material by using predetermined criteria. This objective often is achieved by the direct comparison of mass spectral features of an unknown with either a concurrently analyzed standard or an archived library standard mass spectrum generated with similar instrumentation and conditions. For high-resolution accurate mass spectra, confirmation of identity may include the verification of elemental composition. Characteristic fragmentation patterns obtained under consistent conditions also can be used for the confirmation of identity by providing a fingerprint that indicates functional groups, substituents, and structural differences or modifications. The importance of chromatographic separation (i.e., demonstration of retention characteristics that are similar to a standard) also should be considered as part of an overall identification confirmation. The importance of chromatographic separation is particularly evident with the identification of various structural isomers or enantiomers for which MS alone may not provide sufficient distinction.
The second category comprises true unknowns or compounds for which no or very limited information about structure or background is available and presents the most complex cases for spectral interpretation. True unknowns are encountered less frequently and often necessitate the combination of multiple spectral procedures in order to achieve a complete structural elucidation. For true unknowns, any relevant information (e.g., origin, preparation, matrix, solubility, other compounds if the sample is a mixture) can aid interpretation.
The first step in the interpretation of mass spectra is the determination of monoisotopic mass. This peak is highly dependent on the ionization procedure employed, analytical conditions, and characteristics of the molecules from which the ions are formed. There are prominent differences between EI and API mass spectra, which, for purpose of this discussion, generally result from hard- and soft-ionization procedures, respectively. EI forms primarily an odd-electron radical molecular cation, M+·, and API and other soft-ionization procedures typically produce characteristic even-electron protonated [M + H]+ or deprotonated [M H] molecules.
The Nitrogen Rule can be useful in identifying the molecular ion peak: it states that an odd-electron ion containing either no nitrogen or an even number of nitrogen atoms will be observed at even nominal mass and that an odd-electron ion with an odd number of nitrogen atoms will appear at odd nominal mass because nitrogen has an even integer mass (14) and an odd valence (3). This rule holds for all compounds that contain carbon, hydrogen, oxygen, nitrogen, sulfur, and the halogens, as well as phosphorous, boron, silicon, sulfur, and alkaline earth metals. Applied to even-electron ions (e.g., [M + H]+ or [M H]), the Nitrogen Rule states that an even-electron ion containing either no nitrogen or an even number of nitrogen atoms will be observed at odd nominal mass and that an even-electron ion with an odd number of nitrogen atoms will appear at even nominal mass.
The formation of molecular adducts can assist with determining and verifying mass in both positive and negative ionization modes. Some common adducts observed in positive ion mass spectra are [M + Na]+, [M + K]+, and [M + NH4]+ adducts. Adducts with other inorganic or organic cations, such as Li, Ag, Cs, H2O, acetonitrile, methanol, isopropanol, and small organic protonated or quaternary amines, also can be generated. Adducts present in negative ion mass spectra include those formed with Cl, Br, and formic or triflouoroacetic acid. Multiple adducts are not uncommon in either positive or negative ionization mode mass spectra from API and other soft-ionization procedures and can include combinations of protonated or deprotonated molecules accompanied by additional protons, multiple inorganic or organic cations, or anions. For some applications, it may be desirable to deliberately produce adducts by the modification of the sample solution, matrix, or mobile phase to induce characteristic fragmentation or to denote components in a mixture with common or unique structural features. Dimeric or polymeric analyte ions [2M + H]+ or [2M H] may be observed respectively in positive or negative ion API mass spectra, often with high analyte concentrations. Analogous dimeric adducts with Na, K, and other inorganic cations also can be formed and observed and can provide verification of the MW. Overall, adduct formation and ultimately the presence of adduct ion peaks in mass spectra are highly dependent on ionization procedure, ionization source conditions, type of mass spectrometer or analyzer, sample matrix, mobile phase composition, and the analyte itself.
The relative intensity of mass spectral peaks can provide information about molecular and ion structure. The most intense peak in a mass spectrum is referred to as the base peak and represents 100% relative abundance in normalized mass spectra. In EI mass spectra, the base peak may or may not represent the molecular ion. However, with API and softer ionization procedures, [M + H]+ or [M H] often appear as a prominent peak in the mass spectrum. The relative intensities of peaks in EI mass spectra, and to some extent in API and other mass spectra, can indicate ion stability.
5.3 Elemental Composition and Structure
Each molecule has a specific elemental formula or composition that by itself does not suggest anything about the structure of the molecule, because completely different molecules can have identical empirical composition. The mass spectrum of an analyte indicates its monoisotopic mass, but the larger the mass, the greater the number of possible empirical formulae or combinations of atoms that could produce that same nominal m/z signal.
5.4 Isotope Patterns
The relative intensity and position of isotope cluster peaks in a mass spectrum provide orthogonal information to the m/z data. Isotopes represent variants of a given element with the same number of protons and a different number of neutrons. The number and relative abundance of isotopes are unique to each element and together comprise the isotope pattern for a particular molecule in a mass spectrum. The relative abundance of each isotope also is a function of the number of each element in a molecule. Natural isotopic abundances depend on the source or provenance of a material and are influenced by variations of the isotopic abundances of its constituent elements. Because a unique elemental formula is consistently indicated by each peak coinciding with the m/z and relative abundance of each isotope in a molecular formula, the resulting isotopic fingerprint can be a powerful tool for the prediction of elemental composition.
Chlorine and bromine are examples of elements that produce prominent and easily recognizable isotope patterns in a mass spectrum. Chlorine atoms are characterized by the isotopes 35Cl:37Cl (approximately 3:1) and bromine atoms by unmistakable doublets (79Br:81Br = approximately 1:1) that enable easy identification of ions containing these atoms in a mass spectrum. Combinations of multiple Cl or Br atoms in a molecule yield characteristic isotope peaks that differ by two units and have relative abundances that can be calculated. For some types of HRMS (TOF in particular), the relative intensities of the isotopic cluster can be carefully defined. Because the relative intensity of the isotope cluster is exactly a function of the elemental composition of the molecule (that is easily calculated), isotope fitting of experimental data to theoretical intensity can help provide the correct formula.
Figure 5 presents the isotopic pattern in the molecular ion region for n-butyl benzenesulfonamide collected using ESI with different mass spectrometers, each having different mass resolution. With nominal mass (i.e., quadrupole mass spectrometer) resolution, one can clearly see the A + 1 (m/z 215) and A + 2 (m/z 216) peaks of the [M + H]+ ion at m/z 214. The major contributor to the A + 1 peak results from the occurrence of versions of the molecule containing 13C, and the major contributor to the A + 2 peak, is naturally occurring 34S. With a higher-resolution mass spectrum, the more accurately calculated relative abundances of the A + 1 and A + 2 peaks could be used to estimate the number of carbon and sulfur atoms in the molecule, respectively. An examination of the FT–ICR mass spectrum illustrates the additional isotopic information that can be obtained under even higher-resolution conditions. This spectrum clearly shows that the A + 1 peak, seen as one peak with the other two mass spectrometers, also has contributions from other isotopes. At resolutions afforded by ICR or orbital-trapping mass spectrometers, the various contributors to the A + 1 peak can be observed, and one would get a more accurate estimate of the atoms present.
+'&img=../../images/v38332/c1736-fig5.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 5. Partial ESI mass spectrum of n-butylbenzenesulfonamide under different mass resolution conditions.
Examination of isotope peaks also can reveal information regarding the charge state of an ion. For example, the difference between the A and the A + 1 isotope peaks is 1 m/z for a singly charged ion, a mass difference of 0.5 m/z denotes a doubly charged ion, and a mass difference of 0.3 m/z denotes a triply-charged ion. Thus, the correct charge state can be determined when only one peak or one set of isotope peaks is present, which supports the determination of MW and elemental composition. Because accurate mass and isotope fitting are orthogonal data that each separately can greatly reduce the number of possible empirical formulae for a given nominal mass, the information used together can reduce experimental data to a single or limited set of possible choices.
5.5 The Monoisotopic Ion
The monoisotopic ion is an important feature of a mass spectrum, and recognizing this ion is valuable in determining the elemental composition of a molecule or ion, particularly with accurate mass measurements. The monoisotopic mass of an ion or molecule is the exact mass calculated using the mass of the most abundant naturally occurring isotope of each constituent element. Therefore, the monoisotopic ion is represented by the isotopic peak composed of the most abundant isotopes of its elements. Importantly, MS observes the monoisotopic mass of the analyte, not its molecular weight (i.e., the average including contributions from heavy isotopes). Recognizing the monoisotopic ion peak can be straightforward, and some singly charged ions and elements present simple or easily recognizable isotope patterns. However, the combination of multiply charged ions and multiple elements that collectively contribute to complex isotope patterns occasionally can make determination of the monoisotopic ion peak quite challenging, particularly with polymers and biomolecules. In such cases the benefit of instrumentation that offers higher resolving power is readily apparent.
5.6 Fragmentation
Fragmentation of an organic molecule in a mass spectrometer and examination of the ions produced in the fragmentation process can provide detailed information about unique structural features such as functional groups, substituents, and connectivity. Fragmentation generally is the dissociation of an activated analyte ion. Observed fragmentations are quite diverse and can range from simple events such as loss of water or ammonia to highly complex pathways that involve adduction, multiple rearrangements, and atom migration. In MS, fragmentation is highly dependent on the type of instrumentation and operating conditions. Primary considerations include ionization source, collision zone conditions, and type of mass analyzer. To some extent, controlling these factors permits the selection and optimization of conditions that are conducive to generating desired or structurally diagnostic fragmentation.
EI fragmentation results entirely from unimolecular dissociation that occurs in high vacuum. Formation of a radical molecular ion (M+·) in an electron beam (typically 70 eV) is accompanied by redistribution and accumulation of energy into different vibrational modes, and as a result the molecular ion may undergo fragmentation. Fragmentation typically occurs by bond cleavage and produces an even-electron cation and a neutral odd-electron radical. The converse also can occur, albeit less frequently: an odd-electron radical cation is observed with an even-electron neutral fragment loss. Fragment ions can further dissociate to generate additional fragments. Generally, fragmentation of odd-electron ions may produce either odd-electron or even-electron ions, but even-electron ions fragment to other even-electron ions. The probability of cleavage of a particular bond in a molecule can be correlated with bond strength and the relative stability of the resulting fragment ions. Some general principles apply with regard to the presence of fragment ion peaks observed in EI mass spectra and can assist with interpretation. These principles have been thoroughly discussed in numerous texts on the topic and are not discussed here.
API and soft-ionization procedures predominantly produce ions such as [M + H]+ or [M H] that exhibit limited fragmentation. API and other softer ionization procedures achieve fragmentation by activating ions in a variety of ways, depending on the type of instrumentation.
5.7 Biomacromolecules
Structural elucidation of biomacromolecules using MS offers a number of challenges that are not common among traditional drug substances described previously in the chapter. To start with, biomacromolecular therapeutics encompass many compounds with very different chemical properties that range from proteins and peptides to a variety of glycoconjugates, lipids, and even DNA and RNA complexes. To further complicate the matter, the molecular mass of biomacromolecules can range from under 1000 in the case of some bioactive peptides and lipids to well over 100,000 for vaccines, antibodies, and heparin complexes. Furthermore, many of these biologically derived substances exhibit considerable heterogeneity and thus may not be characterized as a single drug substance but instead as a mixture that is monitored for consistency from batch to batch using a variety of analytical methods.
This section provides guidance about MS characterization of biomacromolecules. Because of the complexity of this broad class of compounds, this section focuses on a few types of compounds to illustrate the benefits of MS, then comments briefly about how MS can be implemented for other biomacromolecular compounds.
5.7.1 peptides and proteins
Details of the process for characterizing proteins and peptides are provided in Biotechnology-Derived Articles—Peptide Mapping 1055 and are only summarized in this section to provide context.
5.7.1.1 Spectral interpretation:
Mass spectral evaluation of proteins and peptides typically encompasses MS data collected on the intact compound(s) to establish the overall mass of the protein followed by tandem MS to generate information about the primary amino acid sequence of the protein or to characterize sites of protein modification. Interpretation of fragmentation spectra to generate the sequence information is a critical component of this approach, and thus some guidelines for the spectral interpretation of peptide fragments are provided in 5.7.1.2 Database searching. Primary and secondary MS-based approaches have emerged as the standards for sequencing proteins by fragmentation and are known as bottom-up and top-down sequencing.
5.7.1.1.1 Bottom-up—
This approach encompasses fractionation of individual peptides generated by proteolytic digestion and sequencing of the peptides by tandem MS. The complete protein sequence then is recapitulated by piecing together the individual sequences. To get complete coverage of the protein, it is necessary to do separate digestions with multiple proteases with different cleavage specificities to get overlapping sequence coverage to rebuild the overall sequence. This approach is currently the most common MS-based approach used to characterize protein therapeutics.
5.7.1.1.2 Top-down—
Advances in MS-based technologies and particularly ECD and ETD fragmentation offer another approach for sequencing proteins, whereby the intact protein is fragmented to generate sequence information without prior need to digest the protein into smaller peptides. Current challenges with this approach include the need for purified proteins; difficulty in getting complete sequence coverage, particularly for larger proteins; the need for considerably larger quantities of proteins than for the bottom-up approach; and a requirement for a high-resolution instrument with ECD or ETD capabilities. However, during the development of proteins therapeutics the availability of large quantities of purified protein is a likely scenario, and with the continued advancement of MS technologies, top-down proteomics may offer a direct option for characterization of some proteins therapeutics in the future.
5.7.1.2 Database searching:
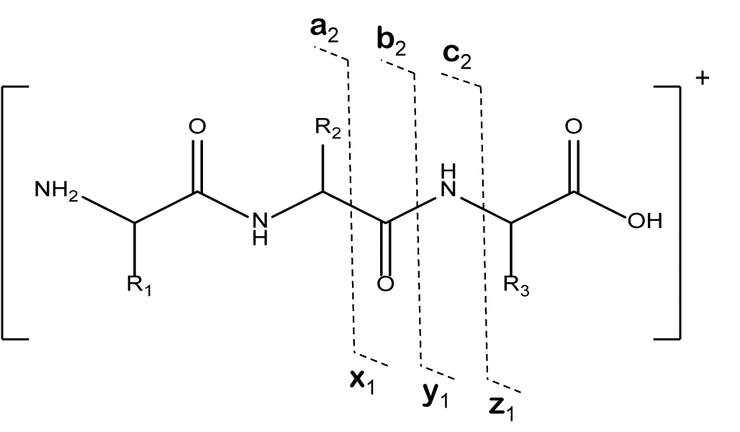
Because of progress in whole-genome sequencing and the computing power of predictive algorithms for the annotation of all possible genes, mRNAs, and proteins from genomic information, the characterization of unknown protein and peptide therapeutics rarely requires complete de novo sequence evaluation. Instead, the compound can be initially identified by digestion and sequencing by peptide fragmentation. This is possible because various fragmentation strategies (e.g., CID and ETD as described previously) produce gas-phase fragment ions from protonated peptides that represent consistent cleavage positions along the peptide backbone. The Roepstorff nomenclature for general peptide fragmentation along the peptide backbone is shown in Figure 6A. Peptide fragments that maintain a charge on the C-terminus are the x, y, or z ions, and those that maintain a charge on the N-terminus are known as a, b, or c ions. By optimizing the fragmentation energies and the collision gas to target only a single fragmentation event per protonated peptide molecule, analysts can use the collective fragmentation spectra from a given peptide to produce a set of fragment ions from the same series (i.e., b ions or y ions) that vary by the residue mass of each amino acid. In this manner they can derive the sequence. A schematic of a peptide fragmentation profile with a complete set of y ions is shown in Figure 6B. The complete set of y ions allows the sequence to be determined from the mass difference between the ions that correspond to residue masses for each amino acid. With the development of complete databases of many genes, transcripts, and proteins, a variety of commercially available computer algorithms have been developed to match the fragmentation masses with protein sequences in databases, along with false discovery rates and confidence probabilities. As a result, confirmation of protein sequences for known proteins can be fully automated for data collection and evaluation.
+'&img=../../images/v38332/c1736-fig6a_2s37.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 6A. Peptide fragmentation nomenclature.
+'&img=../../images/v38332/c1736-fig6b_2s37.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 6B. Peptide fragmentation sequence.
5.7.1.3 De novo sequencing approaches:
When a protein is completely unknown, MS-based fragmentation can be used to generate sequence information as described. The primary challenge is the inability to always get complete, unambiguous sequence information for some peptides by MS fragmentation because of the inability to assign all amino acids from a given fragmentation spectrum without a template of known sequences. In cases such as these, a combination of chemical sequencing using Edman degradation and MS may be necessary. Alternatively it may be necessary to digest and sequence the protein with multiple proteases in order to generate overlaying sequences to ensure the reliability of the overall sequence assignments.
5.7.2 glycoconjugates
The analysis of drug substances in this class is discussed in Glycoprotein and Glycan Analysis—General Considerations 1084. Thus this section addresses only the application of MS to the characterization of protein glycosylation.
Many biopharmaceuticals are glycoproteins for which glycosylation plays critical roles in maintaining function, stability, and solubility. The most common glycosylations are N-linked and O-linked glycosylation in addition to other glycosylation modifications such as GPI anchors. N-glycosylation refers to glycan modification on the asparagine side chain, and O-glycosylation occurs to the side chains of serine and threonine. MS analysis of protein glycosylation can be performed on three different levels: intact proteins, peptides, and glycans. This section provides a brief overview of glycoprotein analysis.
5.7.2.1 Intact protein analysis:
For proteins with small size or relatively simple glycosylation, direct MS analysis of intact protein provides a mass profile that reflects the protein's primary sequence and major modifications, including glycosylation. This type of experiment usually is performed using TOF analyzers or analyzers with higher mass resolution equipped with ESI or MALDI ionization. MALDI forms mainly singly charged ions and requires MS analyzers with a wide m/z range for measurement of intact protein mass (e.g.,TOF). ESI–MS is commonly used, because it leads to the formation of multiply charged protein ions in the m/z range of 700–5000, resulting in better mass accuracy in protein mass measurement (<100 ppm when coupled with a high-resolution mass sepctrometer). MS analysis in many cases can resolve protein peaks by their glycoform distribution because of the mass differences (e.g., hexose 162 Da, HexNAc 203 Da, and N-acetyl neuraminic acid 291 Da). The mass information can lead to identification of the monosaccharide composition of the glycoforms. The intensity of the intact protein peaks can be used to roughly estimate the glycoform distribution on the protein. For monoclonal antibodies modified mainly by neutral N-glycans, quantification of glycosylation distribution by intact protein analysis appears to be comparable with measurement using glycan separation methods. Antibodies containing multiple glycosylation sites can be enzymatically cleaved into major domains before MS analysis to gain more specific information about the position of the glycosylation on the intact protein.
5.7.2.2 Peptide analysis:
For proteins with complicated glycoforms and multiple glycosylation sites, site-specific glycosylation information usually is desirable and is achieved by using a bottom-up proteomics approach via analysis of peptides generated by enzymatic digestion. The peptides are separated by reversed-phase LC followed by ESI–MS/MS analysis. MS–MS fragmentation can be performed using CID or ETD. In CID–MS/MS, the peptides can be identified to be glycosylated peptides by the detection of oxonium ions in the peptide MS–MS spectra [e.g., m/z 163 (hexose), 204 (HexNAc), 292 (sialic acid), and 366 (hexose-HexNAc)]. However, CID–MS/MS spectra of glycosylated peptides usually are dominated with fragmentation from the glycan with minimal fragmentation between the amino acid residues of the peptide backbone, thus hampering the ability to identify the protein sequence. ETD–MS–MS spectra of glycosylated peptides contain mainly fragmentation between the amino acids (e.g., c- and z-type peptide backbone cleavages), while the attached glycans remain intact. ETD–MS–MS provides information about peptide sequence, glycosylation site, and the glycan mass of the glycosylated peptides. CID– and ETD–MS–MS may be used alternatively in the same LC–MS analysis of complicated protein digests, which may provide the information necessary for identification of sequence, glycosylation sites, glycan mass, and glycan branching. O-glycosylated peptides also can be analyzed by using similar approaches. Peptide glycosylation can be identified by observation of oxonium ion such as m/z 204 in CID–MS–MS, and the peptide sequence is identified by ETD–MS–MS when the O-glycan moieties remain attached to the peptide.
5.7.2.3 Glycan analysis:
N- and O-glycans can be enzymatically or chemically released from proteins followed by MALDI–MS or ESI–MS analysis either directly or after derivatization with chemical tags for enhancement of their ionization.
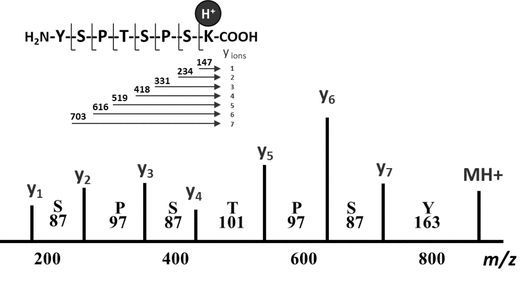
The direct MALDI–MS analysis of released glycans can be performed in both positive and negative mode for the detection of neutral and acidic glycans, respectively. The commonly used matrices include 2,5-dihydroxybenzoic acid (DHB) and 2¢,4¢,6¢-trihydroxyacetophenone. The neutral glycans usually are detected as [M + Na]+ in positive ion MALDI–MS. ESI–MS analysis of glycans suffers from poor ionization of the glycans and thus are used only following chemical derivatization. For example, derivatization of released glycans from glycoproteins can be performed on the reducing end of glycans by reductive amination using fluorescent tags such as 2-aminobenzamide. The labeled glycans are analyzed using hydrophilic interaction chromatography followed by ESI–MS analysis in positive ionization mode. The molecular mass of the glycans can be determined, and subsequent MS–MS analysis can provide structure information about the glycans. Permethylation is another important glycan derivatization method that neutralizes acidic glycans and allows both neutral and acidic glycans to be detected by MS in positive ion mode. MS/MS spectra of glycans contain ions from two major types of cleavages, as illustrated in Figure 7. Glycosidic cleavage occurs at the bonds between two sugar rings and results in B, C, Y, and Z ions, which provides important information regarding monosaccharide sequence and glycan branching. Cross-ring cleavages (shown in Figure 7 by dashed lines) involve rupture of two bonds on the same sugar ring and lead to A and X ions that assist in identification of the type of sugar linkages in the glycans. With increasing understanding of glycan fragmentation, the identification of glycan from its MS–MS spectrum now is possible via database searches using recently developed computational algorithms.
+'&img=../../images/v38332/c1736-fig7.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 7. Nomenclature for carbohydrate fragmentation. Source: Adapted from Domon B and Costello CE (1988). Glycoconj. J. 5, 397–409.
5.7.3 vaccines
Vaccines present a unique challenge because often they include complex mixtures of proteins, polysaccharides, and lipids in the form of intact bacteria or viruses, outer membrane vesicle preparations, or combinations of recombinant proteins. General information regarding bacterial vaccines is available in Vaccines for Human Use—Bacterial Vaccines 1238. Quality control of such mixtures typically relies on characterization by SDS–PAGE, SEC, amino acid or monosaccharide compositional analyses, RP–HPLC, and other analytical methods. However, to date MS has not been used as a primary QC method even though it is frequently used for confirmation of identity and characterization of impurities. Because of the many advantages of MS described in this chapter and its widespread use in the characterization of biotherapeutics and vaccines that contain complex protein mixtures, MS and MS–MS offer the ability to identify product and impurity simultaneously, and likely will be adapted in routine QC environments in the future. Additionally, when a complication occurs (e.g., discrepancy between assays, activity changing after formulation, or posttranslational modification), the quantitation of the recombinant protein (within a complex proteome) or key antigen in a complex protein vaccine via a highly specific procedure such as MS may be desirable. The use of isotope dilution MS–MS in combination with multiple reaction monitoring scans can provide highly specific and accurate quantitation of protein in complex samples.
5.8 Monograph Methods for Qualitative Identification
The discussions in this chapter and in Mass Spectrometry 736 covering the qualitative capabilities of MS provide strong technical justification for its use as part of a compendial identification strategy for a wide range of molecular classes. The structural information content (e.g., molecular mass, empirical formula, structurally significant fragmentation, and characterization of biomolecular modifications such as glycosylation) has been and will be an integral part of identification strategies. One or more specific aspects of mass spectral information can be combined to increase the specificity and reliability of the proposed test. Development of the MS portion of the compendial identification test should strive to provide unique and complimentary information for use with other identification tests intended in the monograph. Method development also should consider the availability of a reference standard for successful mass spectrometric identification.
6. QUANTITATIVE ANALYSIS
MS is useful for the quantitative determination of actives or impurities in a drug substance or a drug product because of its selectivity and sensitivity. The former typically comes into play when target compounds reside in complex matrices (e.g., natural or formulated), and the latter is a benefit when low-level impurities must be measured. Selectivity is achieved in MS-based methods by the combination of well-characterized attributes of the target compound, including molecular mass, intrinsic ionization characteristics, and fragmentation with additional data such as chromatographic retention time. In addition to chromatographic selectivity, use of a chromatographic inlet helps isolate the target compound from other chemical entities within the sample that could create problematic artifacts (e.g., ionization suppression) if they were introduced concomitantly.
This general approach to sample analysis (i.e., chromatography coupled with MS) typically introduces the target compound into the mass spectrometer as a well-defined peak (ideally, Gaussian) that can be reproducibly integrated for optimal quantification. While other forms of sample introduction (e.g., flow injection and direct-insertion procedures such as MALDI) potentially can be employed and may be necessary in some applications, chromatographic introduction systems are more common and are preferred for MS quantification.
Although various mass spectrometer types can be used, the quadrupole and triple quadrupole, in particular, are most commonly employed for quantitative analysis. These instruments provide a combination of specificity, sensitivity, stability, and linear response, which is essential for accurate measurement of target compounds, particularly within complex formulations. For these reasons, the balance of this quantification discussion emphasizes the use of chromatographic separations in concert with quadrupole MS detection.
6.1 MS Quantitation Overview
Employing a chromatographic inlet, analysts introduce a sample solution via injection onto an appropriate chromatographic column, and a flowing carrier stream (gas or liquid mobile phase) advances the target compound(s) and other matrix components through the column at various rates that are determined by strength of the carrier (gas characteristics for GC; organic, along with mobile-phase modifiers, for HPLC), temperature, and affinity for a stationary phase (see 621). Pressurized or near-atmospheric-pressure chromatographic effluent is passed through an appropriate interface into the vacuum environment of the mass spectrometer. In this manner, chemical components within the sample enter the mass spectrometer as chromatographic peaks that are subject to ionization as defined by the chosen ionization mode and according to the propensity of each chemical to become ionized under those experimental conditions.
In the case of single-quadrupole MS detection, the mass analyzer is set to pass to the detector an ion with an m/z value that is characteristic of the target compound. In this selected ion monitoring (SIM) mode, a chromatographic profile for a single m/z value is created. This overall process yields a chromatographic peak at the retention time expected for the target compound. When integrated, the area under this peak leads to a measure of target compound concentration in the original sample. Accuracy in an MS-based quantitative assay is achieved by the proper use of a well-characterized reference standard and, ideally, an appropriate internal standard, as described in 6.2.1 Internal Standards.
For many analyses, the combination of chromatographic separation and SIM MS analysis will provide sufficient selectivity to yield an analysis, which is sufficiently free of chemical interference to meet the measurement need. However, in some situations, it may be necessary to utilize the additional selectivity provided by tandem MS detection that commonly is provided by a triple-quadrupole instrument. The need can be particularly acute in Category II analytical procedures for which a trace impurity must be measured within a complex formulation.
The relative benefit of MS/MS detection is illustrated in the extreme example presented in Figure 8. The antitussive dextromethorphan (Dex) is present in a plasma extract at 100 pg/mL. This extract is first analyzed by LC–MS using ESI–SIM at m/z 272 (protonated molecule). In this experiment the SIM chromatographic peak corresponding to Dex is barely discernible above the relatively high background noise. Alternatively, a triple quadrupole can be employed to set up a selected reaction monitoring (SRM) detection scheme for added selectivity. SRM is a special case of product ion MS–MS in which the second mass analyzer is tuned to pass only a selected fragment ion corresponding to Dex (m/z 147 in this example). A marked enhancement in selectivity, and therefore signal-to-noise ratio, is evident and typically leads to improved performance for quantitative analytical methods.
+'&img=../../images/v38332/c1736-fig8.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 8. MS (SIM) versus MS–MS (SRM) for quantitative methods.
6.2 Quantification Considerations and Procedures
Methods suitable for Category I and Category II applications require the use of a reference standard for each target compound. Quantification of a target compound in an appropriately prepared test sample is achieved by relating its MS response (e.g., SRM-derived chromatographic peak area) to that of similarly prepared reference standards spanning the required concentration range (see Mass Spectrometry 736, Validation and Verification of Mass Spectrometry Analytical Procedures).
6.2.1 internal standards
For optimal accuracy and precision, internal standards should be employed at a consistent concentration in both the test samples and reference standards. Internal standards compensate for small sample-to-sample differences in dilution volume or loss of material during sample processing. They also compensate for analysis-to-analysis fluctuations in MS response or sensitivity drift over the course of batch analyses, both of which occur in all MS-based methods. In practice, selective detection schemes are set up to monitor both the target compound (analyte) and its corresponding internal standard. The key instrumental measure for each standard and test sample is, therefore, a ratio of the analyte-to-internal standard chromatographic peak areas. Internal standards can be either structural or stable-isotope-labeled analogs of the target compound. The former may be adequate and often has the advantage of lower cost and availability. The isotopically labeled (e.g., 2H, 13C, or 15N) alternative offers superior method performance, because it is essentially identical to the analyte in chemical and physical properties, with the exception of greater molecular mass. The only requirements are that the isotopic labels are not subject to chemical exchange during storage or sample preparation, are retained in the MS ionization process, and provide a sufficient molecular mass shift (typically 3+ Da using multiple labels) versus the analyte to ensure negligible cross-talk (interference) between analyte and internal standard signals (because of natural isotopes and variable m/z resolution across different mass analyzers). In addition, care must be taken to ensure that the internal standard contains inconsequential levels of unlabeled target compound impurities that potentially could bias results. Suitability of the internal standard is confirmed after successful completion of the analytical procedure validation.
6.2.2 data acquisition
For accurate integration of analyte and internal standard chromatographic peaks, the data acquisition parameters (e.g., sampling rate, scan range, or masses monitored) must provide a sufficient number of intensity samples across the peak width. The number of samples may vary depending on method performance requirements (see Table 1 in general chapter 736) and practical considerations related to chromatography conditions, mass spectrometer type, and the number of analytes and internal standards monitored. For example, for single-analyte assays on quadrupole-based instruments, analyte and internal standard SIM or SRM detection schemes are alternately sampled throughout the analytical run. With modern quadrupole instruments, dwell times of 100 ms or less per sampling point are readily achieved and should result in at least eight samples across each peak (this is the suggested minimum for good quantitative representation of the peak).
6.2.3 calibration
A calibration curve is created by plotting analyte-to-internal standard peak area ratios versus concentration for the reference standards analyzed. For the somewhat narrow calibration range required for Category I and Category II measurements, a simple (nonweighted) linear fit is sufficient to define this response versus concentration function. However, 1/x or 1/x2 weighting also can be used, as well as nonlinear (e.g., quadratic) functions, as supported by appropriate and provided validation data.
The calibration concentration range is selected based on instrument sensitivity and the required method performance attributes. In general, the lowest concentration should yield sufficient signal for reproducibility, and the highest concentration ideally should be within the linear response range of the MS system. In extreme cases, the upper end of the linear response range may result from limitations of the mass analyzer or detector (e.g., as with TOF and ion trap MS instruments, but much less so with quadrupole mass analyzers). More often, loss of linearity results from diminishing ionization efficiency at higher concentrations. This is a common consideration with, for example, electrospray ionization where high analyte concentrations may increasingly deplete available ionizing adduct ions, leading to a less-than-proportional increase in detected signal with increasing concentration. This so-called self-suppression phenomenon can be somewhat compensated by the use of a stable-isotope-labeled internal standard that co-elutes with the analyte and, therefore, experiences similar proportional suppression. However, best-method performance is achieved when calibration falls within the linear range.
6.2.4 test sample preparation
The scope of acceptable sample preparation procedures for quantitative MS analysis is broad and depends on many factors, including the analyte chemical class, the composition and complexity of the matrix, the target analyte concentration, and the type of analytical instrumentation available. If appropriate, the preferred approach is simple dissolution followed by internal standard addition and then dilution to a target concentration within the calibration range. This simplicity minimizes the potential for analyte recovery issues and contamination or other procedural errors that may compromise the robustness of more complex methods. Provided sufficient instrument sensitivity, optimal method performance typically is achieved with greater dilution factors (lower analyte concentrations in the prepared sample). This approach not only avoids self-suppression issues but also minimizes the potential for ionization suppression due to co-eluting matrix components.
More complex sample preparation procedures may be required to address issues related to the chemical class or concentration of the analyte, as well as the nature of the matrix. In such cases, the use of an internal standard (stable-isotope-labeled standards in particular) is almost essential to ensure adequate method performance. For best results, the addition of the internal standard should take place at the earliest practical stage of sample preparation (e.g., immediately following test sample dissolution).
For example, procedures such as liquid–liquid and solid-phase extraction may be required to enrich (concentrate) target compound(s) for greater assay sensitivity or for isolation from high levels of ionization-suppressing components within the matrix (e.g., salts or other excipients). Chemical derivatization of certain functional groups of a target compound may be desirable to stabilize the sample (e.g., for GC–MS analysis), improve method chromatographic performance, or enhance intrinsic sensitivity for detection (e.g., for ESI). Although such procedures add complexity, they are acceptable provided the overall analytical procedure can be properly validated.
A special case of complex sample preparation that may be required for quantification of macromolecular compounds such as vaccines and biotherapeutics involves the intentional degradation of the target molecule(s) into smaller entities that are more amenable to quantitative MS analysis. For example, partial degradation of a protein therapeutic may be achieved by hydrolytic (chemical) or enzyme-based (e.g., tryptic) digestion to yield peptide fragments that are characteristic of the protein. These smaller molecules can be analyzed by using principles described in this section (including the use of stable-isotope-labeled internal standards of each targeted peptide), and the results can be correlated to determine the concentration of the original target protein. Further, by measuring multiple peptides derived from the protein of interest, analysts can achieve qualitative (identity) verification of the protein by means of the same method. Of course, this indirect approach usually requires some degree of isolation and qualitative identification of the macromolecule to ensure that the subsequent quantitative measure of the representative peptides is relevant.
7. EMERGING MS APPLICATIONS
7.1 Drug Product Authentication and Contamination Detection
Counterfeit and contaminated or adulterated pharmaceutical materials are a threat to patient safety, consumer confidence, and product security and are therefore of concern to pharmaceutical manufacturers and regulatory agencies. Because increases in the incidence of counterfeited and contaminated pharmaceutical materials have elevated concerns about public health safety and product security, numerous measures have been employed to detect counterfeit and contaminated drug products and to ensure supply chain integrity. Because of its sensitivity and specificity, MS is an effective analytical tool for broad application in the determination of authenticity, source, or contamination of drug products. Approaches include verifying the identity of the active ingredient(s), determining and comparing impurity profiles, and isotopic characterization.
7.1.1 identification and/or verification of the active ingredient
Counterfeit drug products may contain multiple or different active ingredients or no active ingredient at all. GC–MS, LC–MS, or direct-infusion MS procedures can provide a verification of the presence or absence of an active ingredient or the identity of other active ingredients that may be present. Counterfeit drug products containing multiple active ingredients or very high levels of a single active ingredient (superpotent samples) typically are of greater concern to human safety than those that contain little or none. In cases such as these, MS can provide positive, unequivocal proof of identity, as well as quantitative information sufficient to pursue those responsible for producing the counterfeit products.
7.1.2 impurity profiling
Impurity profiles can indicate contamination of a drug product or can distinguish an authentic product from a counterfeit. Impurity profiles serve as fingerprints for a drug substance or formulated product from a particular source. Hyphenated techniques such as LC–MS and GC–MS can be used to resolve and identify process-specific impurities that are characteristic of a particular synthetic route, which can be useful in determining the origin and distribution of counterfeit materials. Variations in the impurity profile of the authentic material and changes in the profile with time and process conditions must be considered when analysts use impurity profiles for identification or as indicators of potential contamination. With suspect counterfeit products, similarity of impurity profiles may not necessarily indicate a suspect sample is authentic. In such cases, additional investigation using orthogonal procedures may be necessary.
7.1.3 isotopic characterization
Stable isotopes of elements such as carbon, nitrogen, hydrogen, and oxygen occur naturally in pharmaceutical products and raw materials. Isotopic ratios principally depend on starting materials and processes used to produce drug substances and drug products and can be highly specific for a given batch. Therefore, isotope ratios can define an isotopic fingerprint for a given material, and observed differences in specific isotope ratios between batches can be used to authenticate materials or indicate potential contamination. Isotope ratio mass spectrometry (IRMS) is a powerful tool for pharmaceutical authentication or detection of potential contamination in drug products and raw materials. IRMS affords a high level of specificity, accuracy, and precision in the determination of certain elemental (e.g., carbon, nitrogen, oxygen, and hydrogen) isotope ratios in drug substances or products. Because specific isotope ratios can be influenced by multiple factors such as location, climate, process differences, and source and quality of raw materials, IRMS is able to provide information that is not only useful in authenticating a drug product but also in indicating the origin of a drug product or material. This is particularly important in determining whether distributed counterfeit drug products obtained at discrete locations originate from the same manufacturing process or when linking a diverted product with its original manufacturing site. Figure 9 illustrates subtle but notable differences between authentic drug product samples from three global manufacturing sites and overt differences between authentic and three counterfeit drug products. Additionally, IRMS approaches authentication from a postproduction perspective and does not involve marking or altering the drug product, as may be required by other pharmaceutical authentication technologies.
+'&img=../../images/v38332/c1736-fig9_2s37.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 9. Bivariate plot of stable isotopic composition (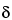 13C versus 15N) of authentic drug product from three global manufacturing sites (Sites 1–3) and three counterfeit drug products (C1–C3).
13C versus 15N) of authentic drug product from three global manufacturing sites (Sites 1–3) and three counterfeit drug products (C1–C3).
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Kahkashan Zaidi, Ph.D.
Senior Scientific Liaison (301) 816-8269 |
(GCCA2010) General Chapters - Chemical Analysis |
USP38–NF33 Page 1637
Pharmacopeial Forum: Volume No. 39(5)