



Add the following:
1. INTRODUCTION AND SCOPE
Many medicinal products are produced through recombinant technology via a host cell (e.g., bacteria, yeast, or mammalian, insect, or plant cell lines). During the manufacture of such products, some amount of non-product, host cell-derived material will inevitably be introduced into the process stream. This process results in a mixture of the desired product and host cell-derived impurities, including host cell proteins (HCPs), and other process-related impurities that will be targeted for clearance through bioprocessing.
Residual HCPs have the potential to affect product quality, safety, and efficacy; therefore, the quantity of HCPs should be low. The product purification processes must be optimized to consistently remove as many HCPs as feasible, with the goal of making the product as pure as possible.
The primary concern with HCPs in biopharmaceutical products is their potential to induce anti-HCP antibodies that could induce a clinical effect in patients. In addition, HCPs may possibly act as adjuvants, which can induce anti-drug antibodies that can affect the safety or efficacy of the drug. A more extensive discussion of immunogenicity and its effect on preclinical and clinical studies is described in USP general chapter Immunogenicity Assays—Design and Validation of Immunoassays to Detect Anti-Drug Antibodies  1106
1106 . HCPs can also have a direct effect on the quality of the product itself. For example, proteolytic HCPs, even in minute quantities, can cleave the desired protein product over time, reducing or eliminating biological potency or altering stability.
. HCPs can also have a direct effect on the quality of the product itself. For example, proteolytic HCPs, even in minute quantities, can cleave the desired protein product over time, reducing or eliminating biological potency or altering stability.
This chapter focuses on HCP immunoassays for recombinant therapeutic products. It does not address products such as vaccines or gene-, cell-, or tissue-based therapies, although the general principles discussed may apply to the measurement of HCPs in these products. The design and validation of immunoassays for HCPs involve unique and significant challenges due to: 1) the wide variety of possible HCPs in medicinal products; 2) the general use of polyclonal antibody reagents to detect them; 3) the lack of exactly matched standards for quantitation; 4) in some cases, a considerable effect from sample dilution effects; and 5) inherent limitations to measure single HCP species.
The chapter includes assay development strategies throughout the product and process development lifecycle, and it describes approaches to demonstrate that the assay is fit for use (e.g., illustrates unit operation clearance of HCPs, lot release). Because of the complexity of HCP immunoassays, careful development and characterization of critical reagents are required, particularly for the immunogen used to elicit the anti-HCP antibodies, the antibody reagent(s), and the assay HCP standard. Because HCP testing is an essential part of process development and product quality control, HCP testing is also discussed in conjunction with regulatory requirements and other considerations for guidance on an overall control strategy for HCPs. A brief outline of the general chapter follows:
1.1 Considerations for Manufacturing, Characterization, and Consistency
Different cell-based expression systems are used to manufacture medicinal products, such as bacteria (Escherichia coli, Pseudomonas fluorescens), yeast (Saccharomyces cerevisae, Pichia pastoris), mammalian cells (e.g., Chinese hamster ovary (CHO), mouse myeloma cell line NSO, and others), insect cells (baculovirus-infected Spodoptera frugiperda cells), and plant cells (tobacco, Arabidopsis, rice). The particular HCP profile is unique and specific to the particular host cells under specific culture conditions and manufacturing processes. HCPs can vary in pI (~3–11) and hydrophobicity, and HCPs display a wide range of molecular weights (from ~5 kDa to at least ~250 kDa), depending on the host cell and manufacturing process used. The number of HCPs in upstream samples can run anywhere from several hundred to more than one thousand proteins, depending on the host cell and culture conditions. Although many cellular hosts have been used in biopharmaceutical manufacturing historically, the most experience has been gained using E. coli and the mammalian cells CHO, NSO, SP2/0, and human embryonic kidney cell line HEK293. The guidance in this chapter draws most heavily from the experience with these expression systems; however, general principles apply broadly to any host cell system.
In mammalian cells, the recombinant protein is typically secreted from the cells into the cell culture fluid (CCF), along with many of the HCPs. However, it has been observed that intracellular protein trafficking may not proceed in a normal fashion in production cultures. For example, proteins usually associated with intracellular organelles, such as lysosomes, may be found in the CCF of largely viable cell cultures, because the clones have been selected for maximum protein export. In addition, as some of the cells die, their soluble, intracellular proteins are released into the CCF. Some harvest operations also lyse cells; therefore, the resulting harvested CCF typically contains both secreted and intracellular HCPs. While this mixture of proteins incubates in the fermenter, additional changes in the HCP population may occur, for example, as the result of enzymatic activity (e.g., proteinases or sialidases).
HCP assays provide important information about the composition of the material entering the downstream recovery process and how each purification step affects HCP clearance. In some cases, HCPs can even bind to, and co-purify with, certain products. Process characterization and validation studies are needed to show which process steps remove HCPs and also to demonstrate the robustness of these steps for consistently removing HCPs. As such, HCP assays are an essential part of purification process development and help ensure manufacturing consistency. Lastly, reproducible and reliable HCP assays may be required to measure residual HCPs remaining in the drug substance (DS) used to make drug product (DP) that is delivered to the patient. HCP levels should be measured in: 1) preclinical lots used in toxicology assessment, 2) all lots during clinical development, and 3) process validation samples from the final manufacturing process. After approval, HCP monitoring may be required as an element of the control system. Subsequent sections of this chapter discuss in more detail the use of HCP assays in process validation and in a good manufacturing practices (GMP) control system.
2. TERMINOLOGY
To help establish a common nomenclature in the literature and with regulatory agencies, Table 1 lists common terms with their definitions (indicating how they are used in this chapter) in addition to synonyms that have been used historically. Note that the term “platform” indicates that the same set of standards and reagents is used within a company to test a variety of products made from the same type of expression system (e.g., CHO cells) grown under similar upstream conditions. In the case of platform HCP assays, the antibodies to HCP are obtained from animals immunized with HCP antigens generated from a common upstream process that is applicable to many products, even if the downstream purifications are different. This approach allows the knowledge from prior products to be leveraged. Justification that an assay is suitable for a new product, using the same expression system and common upstream conditions, is therefore often relatively straightforward.
The HCP immunogen used to generate platform anti-HCP antibodies and used often as the assay calibration standard is, by design, comprised of a broad set of HCPs. In contrast, the qualifier “process-specific” indicates that the immunogen/standard has been prepared from a set of HCPs unique to a given process (either a unique upstream cell culture process or a unique downstream purification process). Process-specific assays are, therefore, limited in their utility, and each must be fully qualified for each process. Process-specific immunogens and calibration standards are, by intent, more narrow and specific to a given process. “Commercially available” assays produced by vendors are often derived from a combination of strains and harvest/purification procedures, and these assays are intended to have a broad application; but these commercially available assays are not specifically designed for a given manufacturer’s proprietary cell line, and users do not have control over reagent availability and lot-to-lot consistency.
Table 1. HCP-associated Terminology
| Term |
Definition |
Historical Synonyms |
|---|---|---|
| Commercially available |
Available to the public for commercial sale; typically a combination of upstream isolates and corresponding antibodies made by the vendor and sold as reagents or kits. |
Generic |
| Platform |
The same set of an HCP standard and antibodies is developed with a company’s proprietary host cell strain and used broadly within a type (e.g., CHO) across several products when the upstream conditions are similar. |
Custom, in-house, proprietary |
| Upstream process specific |
An assay designed from material where the upstream culture process deviates significantly from the platform. This is generally before any purification and may be applied to more than one product if these parameters are similar. |
Custom, in-house, product-specific, proprietary |
| Downstream process specific |
An assay designed from materials where the downstream unit operations are used to enrich the HCP population. This may be applied to more than one product if these parameters are similar. This is rarely used today and is not recommended except for certain products with exceptional downstream processing. |
Custom, in-house, product-specific, proprietary |
| Assay for an individual HCP |
An assay using a standard composed of an identified, single, known HCP and its specific antibody/antibodies. |
Single analyte assay or Custom, HCP-specific |
| Coverage |
Describes the assessment of how completely a population of polyclonal antibodies recognize the population of HCPs. The coverage assessment may be made on the HCP population used as the HCP antigen or from the product production culture. |
|
| Qualification |
Demonstration of suitability of analytical methods (including reagents used in these methods) for their intended application to a given process and in-process samples. |
|
| Protein A/G-affinity purification |
Affinity purification of antibodies with immobilized Protein A or Ga
|
Affinity chromatography |
| HCP-affinity purification |
Affinity purification of antibodies using immobilized HCP (antigen)a
|
Affinity chromatography, immunoaffinity chromatography |
| Null cell |
The cell strain used for production that does not contain the product-specific genetic elements; includes untransfected parental cells and cells transfected with the expression vector but without the product gene. |
Parental, blank, or mock-transfected cell |
| ng/mg |
The numerical quantity (ratio) of HCP per product, where ng represents HCP mass and mg represents the product mass. It is calculated by dividing the HCP concentration (ng/mL) by the product protein concentration (mg/mL). |
ppmb
|
|
a
In some cases, both purifications are performed, typically the protein A/G first, then the HCP affinity.
b
Although ppm has been used historically, it is not advised because this term is used to reflect mass per unit volume for other types of tests. It is recognized that ng is used conventionally as a value derived from interpolation from an HCP standard curve (in units of ng/mL), where the signal is reflective of antibody binding and, unlike the therapeutic protein concentration measurement, does not strictly reflect the mass of HCP that may be present.
|
||
3. HCP IMMUNOASSAY METHODS
Immunoassay methods rely on antibodies that recognize, as broadly as possible, the population of HCPs entering the downstream purification process; therefore, the sandwich immunoassay, designed with polyclonal antibodies, is the workhorse of HCP monitoring and quantitation. This assay format offers a combination of high sensitivity, specificity, throughput, automation potential, rapid turnaround, quantitative results, and low cost per assay that is unmatched by any other currently available assay technology. Other immunoassay formats (e.g., competitive immunoassays) may or may not be suitable, because they lack either the specificity or the sensitivity afforded by the sandwich format. Although these methods result in a single HCP value for a given lot, the number can give greater weight to HCPs for which high-affinity antibodies are present in the reagent(s)—and no or low weight to HCPs which are either not recognized or recognized by low-affinity antibodies in the assay. For these reasons, orthogonal measures of product purity are often needed. More details on these methods can be found in 5. Supporting Technologies for Residual HCP Detection, Identification, and Measurement.
The basic principles and design of immunoassays are discussed in USP chapter Immunological Test Methods—Enzyme-Linked Immunosorbent Assay (ELISA) 1103. The format that is most commonly used for HCP testing is the sandwich immunoassay with detection systems such as colorimetric, electrochemiluminescent (ECL), chemiluminescent, radioactive, or others. Homogeneous immunoassays, including competitive assays, where all of the reagents are combined at once and the binding occurs in a single step without washing, may be problematic due to antigen excess leading to antibody insufficiency issues (discussed later in the chapter); therefore, these formats should be used with caution. The heterogeneous sandwich immunoassay format described in chapter 1103 is generally preferred, because the dynamic range and sensitivity may be reduced in the homogeneous format. The formats, with their advantages and disadvantages, are discussed further in chapter Immunological Test Methods—General Considerations 1102. Data analysis is typically performed with a nonlinear fit of the sigmoidal curve generated by a wide range of standard concentrations, although some analyses may focus on the low end of the curve for greater sensitivity.
3.1 The Assay Development Cycle
Figure 1 illustrates common assay development plans, depending on the reagents available at various stages. Fewer bridging studies are required when: 1) platform reagents are available, and 2) upstream processes are historically consistent. Figure 1A illustrates a scenario where platform methods are not available, and a commercially available assay is used up to the stage of process validation with appropriate assay qualification. For phase III and beyond, either a platform or upstream, process-specific method is preferred. A bridging study should be performed to support assay replacement. If the commercial assay is intended for phase III and post-approval, care must be taken to fully demonstrate that the assay reagents are applicable to the process HCPs. An additional consideration is that the reagents are from an outside vendor over whom the biopharmaceutical manufacturer has less control.
+'&img=../../images/v38332/c1132-fig1.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 1A. If there is no platform assay before product development starts.Figure 1B. If there is a platform assay before product development starts.
Figure 1B suggests that a platform assay can be used through all stages of product development if already available when product development starts. The platform assay should be qualified for each new product. Switching to an upstream, process-specific assay may be necessary for phase III or process validation and beyond, if the cell culture process is significantly changed from the platform process, and may introduce significantly different HCP populations. A bridging study should be performed to support assay replacement.
The limitations of a downstream-specific HCP assay should be considered, because it may be tempting to think that HCP assays are improved by taking the HCPs from null expression through the first column(s) and immunizing animals with a downstream column pool. Historically, the logic was that the immunogen and standard would be enriched in those HCPs most likely to enter the recovery process and be in the final product. This strategy was based on the assumption that, rather than having thousands of irrelevant proteins in the immunogen, only those most likely to be in the process will be present in the immunogen; thus, the resulting assay will be focused on those HCPs of greatest interest. Potential concerns that may arise as a result of this approach are:
-
If only HCPs from a null cell run pool from the first column are used, then later changes during process development (e.g., changes in column run conditions) could invalidate the HCP assay. Thus, process development becomes very restricted and involves the risk of needing to develop multiple HCP assays for slight process changes (or the need to manage the uncertainty).
-
HCPs that co-purify with a product may do so because they bind directly or indirectly to the product protein. A null cell run of a column without the product protein would miss these HCPs.
-
Nonspecific adsorption to chromatography resins is not uncommon and, often, is not a reproducible phenomenon. Compared to the first passage, subsequent passage of the null cell run material over a column will likely produce a different set of HCPs after passage over a new column resin.
3.2 Development and Characterization of HCP Reagents
3.2.1 preparation of antigen/hcp standard
As described in 1.1 Considerations for Manufacturing, Characterization, and Consistency, the total HCP “antigen” is a complex population of proteins; therefore, when generating the HCP antigen/standard reagent, it is important to ensure that: 1) the calibration standard is representative of the cell line and manufacturing process, 2) its protein concentration is accurately quantified, and 3) the immunogen is administered in a way that generates polyclonal antibodies with reactivity to as many different HCPs as possible. The HCP antigen composition should also be comprehensive enough to tolerate normal process manufacturing changes during the life cycle of the product(s).
In addition to the uses above, the HCP antigen may also be needed to prepare the affinity column for purification of the antisera. If affinity purification is used, the quantity of HCP antigen needed should be carefully planned. The amount produced should be large enough to provide sufficient inventory for many years (often 10–20 years); ideally, for the whole life span of the product. However, the antigen preparation process should be performed and documented in a way that facilitates a potential resupply.
Because the HCP assay is used to test DS samples that contain trace HCP impurities, any cross-reactivity of the anti-HCP antibodies with the product may compromise the test method and yield biased results. Therefore, any contamination of the HCP antigen with product must be avoided to prevent the generation of anti-product antibodies.
3.2.1.1 Preparation of HCP antigen from mammalian cells:
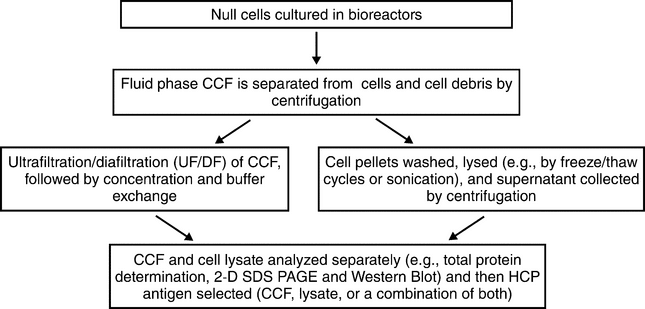
+'&img=../../images/v38332/c1132-fig2.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 2. Example of mammalian HCP antigen preparation.
The first step is to establish a null cell that does not express the product gene, either by using non-transfected (i.e., parental) cells with a common origin to those used to make product alone, or to transfect them with the vector used to create the production cell line without the product coding gene (historically described as a “mock” transfection). The main advantage to the latter is that selective markers (e.g., dihydrofolate reductase or glutamine synthetase) are expressed. Alternatively, antibodies can be raised to selective markers independently. Another advantage is that mock-transfected null cells express selection markers and may be grown in cell culture conditions more closely resembling the manufacturing process. Using pools of mock transfected null lines which have not been cloned allows for the maximum potential genetic diversity and therefore gives the highest potential of generating a broad host cell protein population. However, these cultures, which are seeded with cell pools (not cloned), may demonstrate more variability from run to run. Both approaches [non-transfected (parental) and mock transfected] are commonly used. Both are also subject to the following issues: the cell culture process optimized for the production clone may not lead to similar viability and cell density of the null cells. In addition, in the absence of the stresses experienced by cells producing large quantities of product protein, the null cell lines may exhibit a different HCP profile. These changes in viability, cellular metabolism, and cell density may alter the HCP profile of the antigen, making it less representative of the manufacturing process. More about choosing appropriate HCP antigen preparation conditions is discussed below.
Once the null cells have been established, the HCP antigen is prepared in a bioreactor, cell culture flask, or cell culture bag reflective of the product or process cell culture conditions. A platform cell culture process may be applied, which represents a common manufacturing process for several products (e.g., CHO-derived monoclonal antibodies). Typically, the resulting HCP antigen is minimally processed to ensure a broad HCP spectrum, making the antigen suitable for HCP monitoring of multiple products generated from the platform process. A platform HCP antigen also may be produced by combining several null cell runs with slightly different cell culture conditions (e.g., allowing the culture to remain in the bioreactor for longer periods of time to vary the cell viability). This approach is used to obtain a broader HCP spectrum, and antisera raised against this immunogen may be suitable for HCP detection in products generated from a variety of slightly different processes. These reagents also may increase the robustness of the HCP immunoassay toward process changes.
Alternatively, the HCP antigen may be prepared using an upstream, process-specific approach in which the cell culture process is tailored to mimic the production process for a unique product or specific process. The advantage of this approach may be the high relevance of the resulting HCP antigen to a particular upstream process. As with platform reagents, because no additional processing purification steps are included, the antigen contains a broad range of HCPs, and the antigen will often remain suitable for HCP monitoring after downstream production-process changes. The disadvantage of this approach is that the use of the reagents typically will be limited to a single (or few) product(s) or process(es).
The HCP antigen can be produced from a representative small scale, pilot scale, or production scale run. There are pros and cons for each approach. Pilot or production scale may mimic the actual process best; however, normal variation between cell culture runs may not be reflected if only one run at large scale is applied. Conversely, several small-scale preparations can be pooled and may better reflect the run-to-run variability of the cell culture process. Regardless of the scale, the presence of product should be aggressively avoided, because its presence in the HCP immunogen will compromise the quality of the resulting immunoassay antisera.
After performing the null cell run, the HCP antigen can be prepared from the cell lysate, the concentrated CCF, or as a mixture of both as shown in Figure 2. If the antigen is prepared from CCF, the cells may be harvested at a time when production line harvest is performed or some days later, to allow for some of the cells to lyse and release HCPs into CCF and thereby broaden the HCP spectrum.
To prepare immunogen from cell lysate, the cells are harvested by centrifugation, and the cell pellets are washed and lysed (e.g., by using repeated cycles of freeze/thaw, or high pressure homogenization). Conditions are typically selected to mimic the production process (i.e., to prepare immunogen from CCF, cells and debris are removed by centrifugation, and the CCF is then treated by ultrafiltration/diafiltration). The buffer is exchanged (e.g., into PBS or HEPES), and the CCF is concentrated. The cut-off for filtration should be chosen to minimize loss of HCP, e.g., 10 kDa or less.
3.2.1.2 Preparation of HCP antigen from bacterial cells:
Because there is no obvious approach for overcoming these limitations, in practice the bacterial HCP antigen is usually generated from the lysates of washed cells, using null cell fermentations, that are grown and induced under conditions representing the upstream manufacturing process as closely as possible. As for any HCP antigen, it is necessary to demonstrate that the HCP profile is representative of the manufacturing process before use in assay development. This approach yields the full complement of HCPs expressed and hence is very broad.
Because most animals have been exposed to bacterial antigens and may have pre-existing antibodies to many bacterial cell proteins, it is important to characterize the pre-existing antibody responses, and if appropriate, consider the use of specific pathogen-free (SPF) animals. The presence of significant levels of pre-existing anti-HCP antibodies may confound the analysis of the antibody responses; therefore, it is important to screen preimmune sera from animals for pre-existing antibodies prior to immunization.
Null bacterial cells transfected with the expression vector contain chaperone genes that may be expressed at high levels (e.g., 5% of the total HCPs). Such overexpression of particular HCP impurities might necessitate the development of single-analyte assays for these particular impurities (see 6.1 Assays for Individual HCPs).
3.2.1.3 Characterization of the HCP antigen:
3.2.1.4 HCP standard reference reagents:
Appropriate controls within the assay range may be established to monitor assay performance. Controls may be prepared from independent dilutions of the HCP standard, product samples or intermediate pools, or spiked product samples. However, there is a slight risk that control samples prepared as a dilution from the same HCP standard material can degrade at the same rate as undiluted HCP standard, and degradation of the standard will not be detected. To mitigate the risk, data from several standard curve parameters (e.g., signal, background, slope, coefficient of determination) are often assessed for each assay and used to support HCP standard stability. Using material different from the reference to prepare controls will help ensure that the degradation rate will be independent, and HCP standard degradation can be detected easily.
Assay control charts can be established and used to record reference curve parameters, control sample values, and lot-specific information for the critical reagents, such as labeled HCP antibodies. Acceptable ranges for the controls should be established on the basis of multiple runs (usually 20–30) and may be used as part of routine assay acceptance criteria. In addition, the values for % coefficient of variation (CV) of standard and control replicates are often a part of the assay acceptance criteria. Differences over time in HCP standard curve performance or HCP control levels, or an increase in assay variability, may indicate a lack of HCP standard stability.
3.2.2 preparation and characterization of antibodies
3.2.2.1 Preparation of antibodies to HCPs:
A portion of the prepared HCP antigen is mixed with an appropriate adjuvant (most commonly, Freund’s adjuvant or in combination with incomplete adjuvant) and used for the immunization of animals. Each animal receives an initial priming immunization, followed by multiple (often 4 to 8) booster immunizations, to mature the immune response and generate high-titer, high-affinity antibody responses. Antiserum is collected from each animal before the initial priming immunization (time 0) and at appropriate intervals (usually 10–14 days) after booster doses. Four to six bleeds are usually collected from each animal. However, the duration of the immunization protocol may be extended by increasing the number of booster immunizations, making it possible to collect additional high-titer antisera. Other immunization strategies may also be helpful (e.g., cascade immunization or size fractionation of the HCP immunogen).
Investigators should perform titer or Western blot analyses of the individual test bleeds before antiserum pooling to screen for and remove sera that 1) have low titer, 2) display an immunodominant immune response to a small group of HCPs, or 3) have nonspecific binding characteristics or anti-product reactivity. This evaluation should also be performed on the final antibody pools to demonstrate broad coverage of HCPs and a lack of binding to the therapeutic product. In addition, some laboratories have found it useful to evaluate antibodies for aggregates using size exclusion chromatography as described below. Acid elution conditions may cause some antibodies to aggregate, and multimers of this type, particularly when labeled with either biotin or horseradish peroxidase, may lead to increased assay background. In addition, some laboratories include a final process-scale size-exclusion chromatography step to reduce aggregate levels to low levels (e.g., <5%).
Multiple approaches to the purification of antibodies from antiserum pools have successfully generated antibodies suitable for use in HCP immunoassays. Antibody purification is often performed by Protein A, Protein G, or HCP column affinity chromatography. The application of coated magnetic beads can also provide benefits in some scenarios. The choice of Protein A or Protein G is driven by which animal species was used to generate the antibody. Manufacturers provide standard protocols for these routine antibody purifications. When affinity purifying anti-HCP antibodies, materials such as chromatography resins, which may have been used previously for other products, should be avoided, and additional cleaning cycles should be in place to minimize carryover. Ideally, manufacturers should use a resin that has never been exposed to a DP. Before the affinity purification step, some laboratories also include an initial 50% ammonium sulfate precipitation of the crude antiserum to enrich and concentrate the antibody fraction and thereby extend the performance and duration of use of affinity columns. The use of Protein A or Protein G purification strategies generates reagents in which the anti-HCP-specific antibodies compose a smaller proportion of the total antibody. In contrast, HCP affinity purification strategies selectively purify anti-HCP-specific antibodies from the antisera or antibody-enriched preparations. In this case, the HCP antigens are coupled to an activated resin, and the antiserum is purified over the column following established protocols or the resin manufacturer’s instructions. The purified antibodies may be further purified by size-exclusion chromatography to remove aggregates, then dialyzed, concentrated, divided into aliquots, and stored frozen (typically  70
70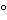 or colder). Neat antisera should also be stored frozen (see 1106 for additional information regarding storage of serum samples).
or colder). Neat antisera should also be stored frozen (see 1106 for additional information regarding storage of serum samples).
Affinity purification of specific antibodies using HCP immobilized on a column requires careful management of the column preparation and use so that it can be reproduced reliably for future purifications. In addition, if reused, appropriate resin storage and regeneration procedures should be evaluated and controlled to avoid degradation during storage. Done properly, it is a reliable, reproducible process that has led to consistent anti-HCP antibodies and HCP immunoassays. After loading the column with antisera, an optimized wash procedure is critical. Because the preparation is a mixture of antibodies with differing affinities, extensive washing may remove low-affinity antibodies. Similarly, loading large amounts of anti-HCP antisera onto the column may result in high-affinity antibodies, displacing low-affinity antibodies if they recognize epitopes that are in close proximity. In either case, the low-affinity antibodies may be removed. In theory, higher affinity antibodies may also be more challenging to elute. Whatever load and wash conditions are initially selected for the antigen-specific affinity purification, these conditions need to be maintained in future production lots of the antibodies. A comparison between these two purification strategies is shown in Table 2. Both approaches are commonly used.
Table 2. Antibody Purification Strategies
| Type of Purification |
Pros |
Cons |
|---|---|---|
| Protein A or G Affinity Column Chromatography |
Robust and reproducible for quickly separating IgG fraction from other serum proteins |
Lower proportion of antibodies specific to HCPs and may have a higher amount of low affinity antibodies |
| HCP Affinity Column Chromatography |
Enriches for specific high affinity anti-HCP antibodies; excludes nonspecific antibodies and serum proteins |
More skill and documentation required to make and maintain resins consistently. Some high affinity anti-HCP antibodies may not be eluted in usable form and may not be recovered |
3.2.2.2 Characterization of antibodies to HCPs:
The quality of the antibody pair (capture and detector) is often evaluated in two ways:
-
Show that the antibody pairs are specific and sensitive in an immunoassay format for the HCPs present in a series of samples taken from the unit operations of a given purification scheme, including the DS. This topic is also addressed in more detail in 4. HCP Immunoassay Method Validation. In concept, the ability to detect and measure HCPs (immunoreactivity) in a series of actual process samples from a given process for a product in development is demonstrated with data showing: 1) sequential clearance as the product is purified, 2) sample linearity throughout a dilution series, and 3) lack of cross-reactivity to product or matrix. The reduction of HCPs during the purification process is determined by clearance factors where the HCP content (in ng/mg) at each step in the process is divided by the amount in the prior step. Typically, starting samples from the cell culture may have HCP levels ranging from several hundred thousand to several million ng/mg, and final products may have HCP levels ranging from <1 to 100 ng/mg (showing many logs of clearance). When these trends are observed, they suggest that the process and the assay are functioning effectively, and the antibodies used in the immunoassay have suitable quality attributes. If these trends are not observed, it may not necessarily reflect a problem with the antibodies, because it is also possible that the purification process is not effective.
-
Show that a broad range of HCPs in the calibration standard is recognized (i.e., that the coverage of the HCP population is adequate). Coverage is evaluated for at least the capture antibodies using 2-D gel Western blots or immunoaffinity fractionation of the total HCP population by immobilizing the anti-HCP antibodies on a column. In addition it is recommended to test the coverage of the detection antibody if different coating and detection antibodies are used. The benefits and limitations of using either 2-D gels or immunoaffinity fractionation to demonstrate coverage is discussed more fully below. Whichever method is used, the extent of coverage must be addressed in qualifying the immunochemical reagents.
HCP coverage evaluations help assess the ability of the antibodies to recognize a wide range of HCPs in the calibration standard and those present in in-process and DS samples. Two methods (2-D gels followed by Western blot analysis and immunoaffinity purification followed by 2-D gel analysis) are in fairly common usage, and both have the limitation that they tend to underestimate the true antigen binding in immunoassay methods but may have value in comparing two preparations head-to-head (or a platform to a commercial reagent). Both procedures use reducing 2-D gel electrophoresis to separate HCPs in the HCP standard or in early process sample(s) (e.g., harvest). Numerical coverage comparisons should be used with caution because of the many method variables and the art required to reproduce results, even with the same reagents in the same laboratory. Because of this variability, the results are best evaluated qualitatively. Comparisons of batches (or sources) of antibodies should be done side by side as much as possible to determine if antibody lots are comparable or if one is superior to another. One approach, the SDS-PAGE/Western, is essentially a comparison of the number of spots present in the immunologically stained membrane versus the number in a duplicate gel (or blot) stained for total protein (e.g., by fluorescent dye or silver; see also USP chapter Immunological Test Methods—Immunoblot Analysis 1104). Differential staining of each antibody preparation may power the analysis, because it can be used to analyze the same 2-D gel. The second approach, immunoaffinity binding/SDS-PAGE, involves comparing the flow through and eluate to the load from the HCP calibration standard (or early process sample) passed over a resin to which the capture antibodies have been covalently immobilized. The resin is washed after loading and before elution to remove nonspecifically bound HCPs. For the purpose of sample comparison, difference gel electrophoresis (DIGE) technology could be helpful. Table 3 lists the advantages and disadvantages of these two coverage methods.
Table 3. Comparison of Coverage Methods
| |
Pros |
Cons |
|---|---|---|
| 2-D SDS-PAGE/Western blot |
Good separation of individual HCPs allows for individual spot counting. |
HCP antigens are denatured, may not represent what is seen in immunological assays (e.g., ELISA). |
| |
High variability—numerical “percent coverage” values vary widely with the same material tested within a single lab and between different laboratories. |
|
| |
Transfer efficiency of a broad range of HCPs difficult to optimize, leading to underestimates due to over-transfer through the membrane or failure to transfer from the gel, dependent on molecular weight. |
|
| |
Matching spots between blots and gels are difficult and not standardized. |
|
| Immunoaffinity binding/ 1- or 2-D SDS-PAGE |
HCPs bind to antibody resin in solution under native conditions similar to the sandwich immunoassay. |
May underrepresent some HCPs if they are bound too tightly and do not elute, resulting in an underestimation of coverage. |
| Analysis of spots in gels does not require immunoblotting and is simpler because the problems of transfer are avoided. |
Preparation of the anti-HCP resin must be done carefully and may be difficult to reproduce. Results can be dependent on resin loading and elution conditions. |
|
| |
Proteins at low concentration that only show up in Westerns will not be detected. |
More information about the appropriate use of SDS-PAGE/Western blot methods and ways to minimize the disadvantages above can be found in the later section 5.2 Considerations for Western Blot Methods.
An example of a suitable use of this approach is shown in Figure 3.
+'&img=../../images/v38332/c1132-fig3.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 3. Left panel: 2-D IEF/SDS-PAGE analysis of representative CHO HCP calibration standard stained with a sensitive fluorescent stain. Right panel: Western blot analysis of the same gel shown in the left panel.
3.2.3 generation and qualification of replacement reagents
When the supply of HCP reagents are depleted or if their performance declines, new reagents may be needed. Often, new HCP antigens and new HCP antibodies are both generated at the same time. In theory, the new set of reagents should be created by following the same protocol that was used for the previous set of reagents to match the performance of the new and old assays as closely as possible and produce consistent data for a given program. In practice, however, it is not always possible (nor advisable) to generate the assay reagents exactly the same way that they were originally made. For example, if the manufacturing process has been optimized over time and the original platform assay reagents are no longer relevant, a careful assessment should be made. When such changes are required, the ideal way to confirm the quality of the replaced reagents is through characterization of antibodies (see below) and demonstrating that they can detect similar or greater HCP log reduction, from harvest to DS, using the same samples tested head to head with the original assay reagents.
When replacing an HCP antigen in which the current production process is essentially the same as the old process, the new HCP antigen should be prepared as similarly to the old as possible. It is important to compare the protein concentrations of the old and new HCP antigens side by side, using the same method to correctly assign the protein concentration for the new antigen and to ensure that the protein concentrations are comparable in the new and old HCP calibration standard. Characterization by 1-D and 2-D SDS-PAGE should also be performed as a side-by-side comparison of the old and new HCP antigens (see above) to be sure that gross changes in protein composition have not occurred.
For antibody replacement, the same animal species should be used, if possible. In cases where a better antibody response is desired, antibodies produced by different species may be evaluated. The selection of the species should take into account all the points discussed in this chapter. A side-by-side comparison of results from process samples using old and new antibodies in a sandwich immunoassay format should be included to demonstrate suitability of the replacement antibodies. Additional characterization by orthogonal methods (e.g., coverage using 1-D and 2-D SDS-PAGE) is recommended, as described above. Assay qualification or validation should also be considered after changing reagents.
To fully understand the effect of replacing HCP critical reagents on assay results requires testing of process samples in bridging studies using multiple samples from harvest, process intermediates from all appropriate purification steps, and the final DS. These studies can be very resource intensive, which underscores the importance of making sufficient quantities of HCP critical reagents at the outset (and minimizing replacement).
3.3 Immunoassay Method Development and Qualifying as Fit for Use
As noted in 1. Introduction and Scope, HCP immunoassays serve two important functions in the development and control of biopharmaceutical manufacturing processes. HCP immunoassays serve as a measure of product purity, and therefore are potentially related to patient safety. They also serve as a measure of manufacturing consistency, and therefore they are reflective of process control. To meet these requirements, HCP assays are often validated and incorporated into the cGMP control system, which includes specific reject limits (in the DS as a Certificate of Analysis (C of A) method or as an in-process control). If the final DS is a conjugated protein, the testing should be done on the protein intermediate (before conjugation). In addition, HCP assays are used to guide process development and are often used to demonstrate robust process performance in the context of formalized process validation activities.
HCP immunoassays are often formatted as a sandwich ELISA, and multiple antibody labeling options are available (see 1103). Depending on the label chosen, appropriate conjugates and substrates are selected; examples are described in 1103. Most proteins can be biotinylated because of their lysine content and, because biotin is small, it is less likely to affect the protein’s binding activity. Because antibodies are large and have many lysine residues, the conjugation ratio of biotin to antibody can be higher than that of smaller proteins. However, it is important to not over- or under-label the antibody (e.g., with biotin or HRP), because this might lead to either less antigen binding or lower signal/sensitivity, respectively. Controlling and defining the labeling stoichiometry (e.g., mole of biotin to mole of IgG) is useful for making future batches consistently. Multiple ratios of any antibody label should be tested and optimized for performance in the immunoassay. Excess unconjugated reagents should be removed, either by dialysis, affinity purification, or by using a suitable desalting column.
Often, the standard is first screened to see whether a dose-response curve can be generated, and if so, in which concentration range of analyte. The results can also reveal a starting point for initial reagent concentrations that can be optimized further. If more than one antibody pair has been prepared, then each is tested to find the best performance (see USP chapter 1103 for more information). The signal (low calibration standard) to noise (background) ratios generated with each antibody candidate can be used to select the best pair. Typically, HCP immunoassays do not always possess a full dose-response curve with both asymptotes; therefore, for the purposes of residual HCP detection, the assay for DS release should focus on the low end of the curve.
3.3.1 qualifying a new manufacturing process with a platform hcp assay
As noted earlier, if the upstream/isolation procedure is matched, then a new product produced in that system can benefit from a platform assay approach that can decrease qualification time and effort. However, in cases where there are variations that occur in optimizing the production cultures and the cell culture design space is not completely defined, it is important to understand whether these variations are significant enough to render the platform assay unsuitable. In these cases, the platform assay should be qualified experimentally to demonstrate that it is suitable for measuring HCPs from the new process. Assay accuracy, precision, sensitivity, linearity, range, and specificity must be evaluated (see 4. HCP Immunoassay Method Validation). In addition, the range of HCPs in the platform HCP standard should be compared to those present in the new process. The antibodies should also be characterized using samples from the new process or product using approaches discussed previously.
The following approaches and concepts may be useful in determining if a platform assay is suitable for a product made with a new process:
-
Conduct a representative, small-scale null cell run grown under conditions used in the new culture process, and determine the protein concentration of the null cell run CCF. Assay the null cell run CCF at the same nominal concentration as the assay HCP standard using the HCP sandwich immunoassay, and compare the dilution curve from the null cell run CCF to the standard used in the assay. If the standard and new null cell run CCF curves are similar (e.g., in shape and amplitude, and the calculated HCP concentrations are within a factor of two), then the new process is considered not different from the platform, and a process-specific HCP assay is not usually needed.
-
With the CCF from the above null cell run from the new process, make a comparison to the HCP calibration standard using 2-D gel electrophoresis. This will compare the diversity of proteins produced by the two processes. However, be aware that new “spots” may appear in the 2-D gels that may not reflect immunochemical differences in the assay, for example, post-translational modifications or limited proteolysis. As such, the appearance of a few new spots or up-regulated or down-regulated spots in the process-specific null cell run is not a basis for invalidating the application of the platform assay. A qualitative assessment (e.g., versus a Western blot) is recommended.
-
Comparison of HCP levels at harvest in actual production runs (CCF samples) with the same product produced in the original versus the “new” process is also valuable. In general, immunoassay results within a factor of two are often considered similar, thereby confirming the platform HCP reagents.
-
HCPs in CCF from production cultures may also be estimated on the basis of total protein measurements. The total protein in the CCF is the sum of product (measured using a product-specific titer assay) and HCPs. Samples from harvests of the manufacturing run cell culture can be dialyzed, as long as they are sufficiently concentrated to make membrane losses negligible, to remove small peptides and amino acids, and then the total protein is determined (e.g., by amino acid analysis). Subtraction of the product concentration from the total protein provides an estimate of the non-product protein, HCP, in mg/mL. This value may be compared with the HCP concentration from the immunoassay. Comparable values (e.g., within 2×) indicate that the HCP platform is confirmed for the new process.
3.3.2 need for new reagents after process changes
Major changes, such as substantial and unambiguous changes in cell culture conditions (e.g., moving from a serum-containing process to a serum-free production process), typically require production of new reagents and development and validation of a new HCP assay. In this example, the HCP reagents created using the old process recognize a number of serum proteins but may not recognize many new HCP proteins in the serum-free process. Because of both the process change and the assay change, different HCP levels will be observed when the same samples are measured for HCP in the old and new assays (i.e., the samples from the serum-containing process may have a high level of HCP measured by the old assay and low level of HCP measured by the new assay, whereas the samples from the serum-free process may have much lower HCP results by the old assay but more with the new assay).
If the assay was developed as a downstream, process-specific assay, changes in the purification steps can also require replacing the assay with a new one that is relevant to the new downstream process. This is the biggest problem with these very specialized assays and why platform-based assays are usually preferred, because they often do not require a new assay when the downstream steps are changed.
3.3.3. sample stability
It is important to demonstrate that the sample handling procedures do not affect the assay results. This is particularly true when comparing results for various intermediate process pools. Occasionally, HCPs can precipitate during freezing or when the sample pH is adjusted from acidic to neutral. In addition, storage at 2–8 or multiple freeze-thaws may result in loss of reactivity. The analyst may need to add specific stabilizing components (e.g., divalent cations) to the samples to ensure stability.
4. HCP IMMUNOASSAY METHOD VALIDATION
HCP immunoassays for product purity assessment typically are part of the cGMP control system, either as an in-process test or on the C of A, and need to be validated appropriately. The validation approach depends in part on the types of samples that will be tested with the method. All validation parameters for a quantitative impurity test are needed when used for the final DS, whereas validation for in-process samples usually focuses on dilution linearity, interference, and precision. Regardless of whether the assay is an in-process or release assay (DS), action limits are valuable as a part of an overall control strategy. Readers are referred to ICH Q2(R1) guidelines and chapter Validation of Compendial Procedures 1225 for general expectations for assay validation, particularly those requirements for cGMP assay validation, and to chapter 1103 for sandwich immunoassays. Although the main focus of this section is on cGMP assay validation, many of the same principles apply to the assays used to demonstrate that process purification steps are suitable for their intended purpose.
This section will focus on those aspects of HCP immunoassays that are unique or require special attention and documentation. HCP immunoassays differ from more conventional immunoassays in that they are multi-analyte assays. There are potentially thousands of proteins in the immunogen and standard, and a correspondingly diverse set of antibodies is produced, yet only upstream CCF will contain a diversity of proteins that approximate the HCP calibration standard. Intermediate pools and final products typically contain progressively fewer of the HCP proteins that are present in the standard. As discussed previously, this is the aspect of accuracy that is compromised by HCP immunoassays, and why the mass unit ratios (ng of HCP per mg of DS) are not a literal “mass” relationship. However, below are common industry approaches that address these limitations as much as possible.
The validation summary below is for the final DS or a penultimate process step, whichever is chosen for the C of A method. It should be noted that for in-process testing, the focus is on method accuracy (spike recovery), dilution linearity, and precision. In the case of accuracy, some samples will have more potential interfering substances (other process impurities or additives) that may need to be confirmed as non-interfering (e.g., residual DNA, leached protein A, virus inactivators, chaotropes, and low pH buffers).
4.1 Accuracy
To assess HCP assay accuracy in the DS, at a minimum the analytical assay standard should be added to appropriate samples and accurate spike recovery demonstrated over the range of the assay. Matrix interference can come from buffer components and product, and the minimum dilution required for acceptable spike recovery (typically between 70% and 130%) needs to be determined for each sample type. Typically, spike recovery experiments are conducted with spikes using at least three and potentially as many as five different levels. Because samples are assayed at multiple dilutions, the investigator may spike and then dilute the sample, or dilute the sample and then spike the diluted samples. Spikes near the quantitation limit (QL) help to evaluate the accuracy and repeatability of the assay near the QL, which is where the measurement is often the most variable. A spike recovery of 50%–200% may be acceptable for a spike at or near the QL.
Although these are minimum requirements for assay validation, they should not be interpreted as demonstrating accuracy for any one specific HCP that may co-purify with the product. To accomplish that, comparison to a standard of that particular HCP species is needed; however, because this HCP is rarely known, this may not be possible. Tips for discerning these situations are provided below in the dilution linearity section and in 5. Supporting Technologies for Residual HCP Detection, Identification, and Measurement.
4.2 Sensitivity and Assay Range
HCP sandwich immunoassays often achieve standard curve sensitivity in the low single digits of ng/mL. For sample protein concentrations where the test dilution is 10 mg/mL, this implies that a sensitivity of about 1 ng of HCP per mg of product is possible. However, this sensitivity may be difficult to achieve for products with lower protein concentrations. It is important to highlight that the reported HCP ratio (ng of residual HCP relative to mg of product) is not really the ratio of masses implied by the unit ng/mg but rather “immunological equivalents” per mg of product.
The first aspect of determining QL is typically determined experimentally for each product in a given sample buffer matrix in spike recovery studies. The minimum recommended dilution (MRD) for the DS should be established by spiking the HCP standard in a sample dilution series. For example, an HCP spike of 10 ng/mL added to each of a series of solutions containing undiluted (e.g., 10 mg/mL) protein and serially diluted DS. Those dilutions in which the spike recovery is 70%–130% (50%–150% is also commonly used for levels near the QL) are considered acceptable. Typically, a proposed DS MRD is tested further to ensure that spike recovery is consistently achieved.
For the second part of the QL determination, the DS is tested at the MRD (if the DS samples have high levels of detectable HCP, a formulation buffer may be used). The QL is generally determined by the analysis of spiked concentrations of HCP and by establishing the minimum level at which the HCP can be determined with acceptable accuracy and precision. This study is typically performed at least three times, preferably by different analysts or on different days. For example, a spike of 3 ng/mL of HCP in a product protein concentration of 5 mg/mL has a QL of 0.6 ng/mg or 3 ng/mL if spike recovery is achieved in, e.g., at least four of six tests or if mean spike recovery criteria are met.
Typically, assays are set up to measure the range from a few ng/mL to >100,000 ng/mL. This range allows the analyst to test a variety of samples at multiple dilutions (see discussion of 4.3 Sample Linearity) and allows for a practical assay that can accommodate in-process pools with highly differing HCP levels. For example, upstream samples may contain >100,000 ng/mL of HCPs and require large dilutions to obtain results in range of the standard curve.
For routine commercial DS manufacture where the protein product concentration is known and the process impurities are well understood, testing at a single dilution may be used for release. The results are reported as the ratio of measured HCP (ng/mL) to the product concentration (mg/mL) resulting in units of ng/mg. When the DS has undetectable levels the results are reported as “less than” the assay QL (ng/mL) divided by the product concentration (mg/mL; e.g., <0.6 ng/mg in the example above). Before setting this target concentration for testing, however, the dilution linearity of the samples should be well understood and a robust manufacturing process established. In the event that the level or species of HCPs vary run-to-run it may be necessary to test each sample at multiple dilutions (see below).
4.3 Sample Linearity
HCP assay nonlinearity is not uncommon for some samples and should be assessed for multiple batches of a given sample type (e.g., from several clinical DS lots or process validation batches) to ensure consistency and proper reporting of results. Because a single HCP (or a few individual HCPs) may co-purify with product, it is possible that particular HCP(s) will be present in excess of the available antibodies in the HCP immunoassay. This is possible in multi-analyte assays where only a limited surface area is available on assay microtiter plates or beads, and because thousands of different anti-HCPs are needed, antibodies to any one HCP will, of necessity, be limited. Furthermore, the relative amount of antibody to each separate HCP is also not controlled, so the binding capacity for each separate HCP differs. For samples exhibiting this behavior, the analyst must dilute the sample into the range of the assay, i.e., to a point where nonlinear behavior is no longer observed because the HCP has been diluted to a concentration where it no longer exceeds the available antibody. In some cases, the sensitivity of the assay becomes limiting, and one never reaches a dilution where the assay result is independent of the sample dilution. In such cases, the highest HCP ratio value (corrected for sample dilution) within the validated assay range should be reported. A detailed example of how to handle nonlinear dilutions arising as a result of antibody insufficiency follows.
Table 4 and Figure 4 show data for three different samples tested in an ELISA that has a range of 1–100 ng/mL. In this example, all samples were diluted initially to 10 mg/mL (the MRD, where spike recovery had been previously confirmed), and then serially diluted using a twofold dilution series to below the assay QL of 1 ng/mL. Whereas Sample 1 (diamonds) dilutes linearly over the full range tested, both Sample 2 (squares) and Sample 3 (triangles) HCP results plateau at higher concentrations of the sample. Saturation of the antibodies reflects antigen excess, and determination of the HCP value is made by diluting the sample into the range of linear dilution; visually estimated as <2 mg/mL for the squares and <1 mg/mL for the triangles. [Note—If large sample dilutions are required to get into the range of the HCP assay, consider making intermediate dilutions to limit dilution-related errors.
| Product |
Sample 1 |
Sample 2 |
Sample 3 |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Conc. (mg/mL) |
HCP conc. (ng/mL) |
HCP ratio (ng/mg) |
% max ratio value |
HCP conc. (ng/mL) |
HCP ratio (ng/mg) |
% max ratio value |
HCP conc. (ng/mL) |
HCP ratio (ng/mg) |
% max ratio value |
| 10.00 (neat) |
49 (neat) |
4.9 |
83% |
20 (neat) |
2.0 |
<20 |
3.2 (neat) |
0.3 |
22% |
| 5.00 |
28.5 |
5.7 |
96% |
16.5 |
3.3 |
54% |
2.5 |
0.5 |
35% |
| 2.50 |
12 |
4.8 |
81% |
10 |
4.0 |
66% |
1.5 |
0.6 |
42% |
| 1.25 |
7.4 |
5.9 |
100% |
7.4 |
5.9 |
97% |
1.1 |
0.9 |
83% |
| 0.63 |
3.1 |
5.0 |
84% |
3.3 |
5.3 |
87% |
0.9 |
1.4 |
100% |
| 0.31 |
1.6 |
5.1 |
86% |
1.9 |
6.1 |
100% |
<1 |
<6 |
NA |
| 0.16 |
<1 |
<6 |
NA |
<1 |
<6 |
NA |
<1 |
<6 |
NA |
| Reported HCP ratio value with guide and n
|
Guide 1 = 5.2 (n = 6); Guide 2 = 5.2 (n = 6) |
Guide 1 = 5.8 (n = 3); Guide 2 = 5.3 (n = 4) |
Guide 1 = 1.2 (n = 2); Guide 2 = 1.4 (n = 1) |
||||||
|
a
Numerical data for three samples (also graphed in Figure 4) show differing degrees of saturation of the response at higher concentrations of product (and HCP impurity) tested. Two guides on how to interpret these data (guides 1 and 2) illustrate how the “linear range” of the assay is limited to the most dilute data points shown in Table 4 and Figure 4. Note—Samples are always diluted until the HCP concentration is below the QL of the assay (in this example the QL is 1 ng/mL). Guide 1: All values within 20% of maximum value are averaged. Guide 2: Values are averaged as long as the CV of the values is <20%, removing less diluted samples first. A third guide might also be used that reports the highest value measured above the QL but the suitability of this approach should be very well demonstrated by method validation.
|
|||||||||
+'&img=../../images/v38332/c1132-fig4.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 4. Twofold serial dilution of the three samples listed in Table 4. HCP concentration results are plotted against the product concentration in the samples, which serves as a measure of sample dilution.
Because measurements near the QL may have higher variability and less reproducibility than measurements farther from the assay limit, usually (if possible) values within the sample linear HCP concentration response range are averaged. The procedure for which data points may be averaged must be specified in advance of running the assay and is typically documented as part of the test procedure. In the example shown in Table 4, two different guidelines are illustrated. Guide 1 averages all values within 20%–25% of the maximum HCP ratio value. Guide 2 averages values only if the CV for the resulting averaged values is <20%–25%. Although their data sets are not identical, both rules are justified, based on the argument that variations of 20%–25% are within the variation seen in assay replicates and reflect the underlying reproducibility of immunoassay methods. In this example, both methods give very similar results. In contrast, the third potential guide, which reports the highest value measured above the QL, would yield results that are at least 10% higher. In addition, the validity of the third guide would depend highly on the quality of the method development.
Homogeneous (one-step incubations without washes between reagent additions) sandwich immunoassays are more prone to antigen excess and resulting dilution nonlinearity problems and may exhibit “high-dose hook effects” in which samples with large amounts of antigen give inaccurately low readings, because the antigen excess prevents formation of the complete sandwich. Suitable use of this format should be confirmed experimentally for each sample type and could be acceptable if the samples are diluted to assay QL and lack of hook effects is confirmed.
Historically, some companies have used spike-recovery data produced in the validation of accuracy to demonstrate linearity. Although this demonstrates the linear dilution of the standard, it does not demonstrate the linear dilution of the HCPs in the samples. For the reasons of antigen excess discussed above, the linear dilution of the standard is a necessary condition but not sufficient to demonstrate linearity of the assay for different sample types.
Finally, although antibody insufficiency is generally believed to be the primary cause of a lack of dilution linearity, other potential causes are: 1) reactivity of the antibodies with product protein, 2) the presence of non-specific antibodies (“sticky” antibodies), 3) reactivity of the antibodies to non-HCP proteins present in samples (such as peptones or BSA), 4) HCP aggregation, 5) HCP standard dilution profile that is not representative of the in-process or final product HCP, and 6) sample matrix interference. Cross-reactivity and matrix interference are discussed below.
4.4 Specificity
Assessment of potential matrix interference in samples is addressed in section 4.1 Accuracy and is primarily used to establish the lack of formulation interference (in the DS) and other impurities that are normally present (for upstream process samples). Another specificity issue to evaluate is the potential cross-reactivity of the anti-HCP antibodies with the product itself. Although this is possible, antibodies to HCPs that cross-react with product epitopes are rarely observed.
In those cases where cross-reactivity is observed, more false-positive assay signals are risks that must be managed. If cross-reactivity is suspected, it should be carefully and scientifically established. In the vast majority of cases, signals in HCP immunoassays are real, indicating the presence of HCP impurities. In particular, instances where the HCP(s) are noncovalently associated with product, and they co-purify, may give the appearance of cross-reactivity. However, because of the tremendous variety of antibodies in a polyclonal anti-HCP pool, when measuring residual HCP in the presence of excessive levels of product, some antibodies may still bind product, yielding a false-positive signal. In this case, it may be necessary to modify the assay or the anti-HCP reagent. If a platform HCP assay is used to test multiple batches (for the same program), and different levels of HCP between batches are observed, this is strong evidence that the HCP-positive signal is real and not due to cross-reactivity.
Evidence for cross-reactivity is often seen in a product that has been purified in a sequence of steps and where the HCP levels are not reduced by typical purification processes. The HCP/product ratio (ng/mg) should be used so that changes in the HCP/product ratio during purification can be compared. First, Western blots of DS are usually probed with anti-HCP antibodies to determine if HCP(s) are noncovalently associated with product. If bands are detected that are unrelated to the product, this suggests that cross-reactivity is not occurring, and further process development may be required if lower HCP levels are sought.
Alternatively, if the anti-HCP is recognizing a product-associated band, this suggests cross-reactivity. Caution should be exercised, however, because the product is denatured (by SDS-PAGE), and binding may be to epitopes not accessible when in the native, solution phase (i.e., in the immunoassay). In this case, a positive Western blot could be an artifact (conversely, a negative Western blot could also miss cross-reactivity, but this is less probable). Regardless, if cross-reactivity is suspected, it can be confirmed and corrected by either: 1) addition of a high-purity product (or related structures, such as human IgG from a different source, for a monoclonal antibody) to block the cross-reacting antibodies in the assay, or 2) depletion of the anti-HCP in the reagents using a product-affinity column made from highly purified product. In either case, it is important to ensure that the antigen used to block or deplete the serum is itself free of residual HCPs. This information is available from the Western blots probed with anti-HCP. If HCP bands are present, this indicates that a higher-purity product may be needed and can be prepared with orthogonal, lab-scale purifications. Sometimes reversed-phase HPLC can be effective in making this material, although this is not useful during product production because of its denaturing properties.
5. SUPPORTING TECHNOLOGIES FOR RESIDUAL HCP DETECTION, IDENTIFICATION, AND MEASUREMENT
Biopharmaceutical manufacturers need an integrated control system that offers assurance of overall product purity and manufacturing consistency. Typically, product purity is demonstrated by using a combination of methods, and this is very much the case for HCPs. In addition to the most common sandwich immunoassay format, other assay types provide valuable complementary information. Tables 5, 6, and 7 provide overviews for commonly used methods for HCP detection, quantitation, and characterization. Table 5 discusses electrophoretic methods; Table 6 discusses Western blot methods; and Table 7 discusses chromatographic and proteomic methods. Gel electrophoresis and immunoblots are also discussed in 3.2.2.2 Characterization of antibodies to HCPs.
Orthogonal assay methods used to provide additional assurance of purity and the lack of HCPs may be used in any of three modes: “detection”, “identification”, or “quantitation”. In the case of detection, orthogonal assays address the question, “Is there any protein other than a form of the product that the immunoassay may have missed?” If this is the case, as knowledge accumulates regarding the identity of the HCP(s), it becomes possible to perform a specific protein risk assessment and, if necessary, generate a standard and separate test for that particular HCP impurity. The orthogonal method may then be used in a “quantitative” mode by comparing the product sample with the appropriate HCP standard for the known HCP. For example, a quantitative HPLC-MS/MS method may be developed, based on monitoring peptides that ionize well from digests of the analytical standard protein and the sample.
5.1 Considerations for Electrophoretic Methods
Three electrophoretic methods that are useful in impurity analysis are 1-D and 2-D SDS-PAGE and SDS-capillary electrophoresis using non-gel sieving (CE-SDS). In all these methods, excess sample is loaded to ensure detection of trace protein impurities. The drawback is that excess product obscures impurities that migrate close to the product and cause interference. However, 1-D gels or CE-SDS may already be part of the GMP control system for monitoring product-related fragments; therefore, extending these methods for HCP monitoring does not involve additional testing. 2-D gels have been used to investigate the purity of the final product and to identify minor spots in the gels associated with either product variants or HCP impurities. The presence of a new band in the gel, or a new peak in the CE-SDS electropherogram, may represent either a new product-related substance/impurity or may reflect the presence of an HCP impurity (“process related”). In either case, an investigation is warranted. If not part of the routine control system, a 1-D gel is still an excellent method for extended characterization of important lots, such as registration lots, and for manufacturing investigations. Gels and CE-SDS are useful for confirming the absence of gross levels of HCPs (should they be present and missed in the immunoassay). Gels are typically stained with a high sensitivity stain, such as a fluorophore or silver. Ideally, the stain should be compatible with in-gel proteolytic digestion and subsequent analysis by mass spectrometry to identify the protein.
Proteins differ in their staining intensity with silver or fluorescent dyes; therefore, only semiquantitative estimates of the amount of protein in a band or spot are possible (e.g., glycosylated forms may not stain as intensely). Nonetheless, because these methods do not rely on anti-HCP antibodies, they provide an important orthogonal measure of purity.
Some proteins may appear in gels as multiple bands or spots that result from post-translational modifications or proteolytic clipping. To the extent that a single gene product is distributed to multiple locations in the gel, this further reduces the sensitivity of gels to detect impurities.
As discussed following Table 7 on proteomic methods, it is often possible to excise bands or spots from the gels. These excised gel fragments may then be incubated with proteases to provide in situ digestion of the protein into peptides. These peptides may then be fractionated and sequenced using LC-MS/MS and compared with databases to identify the origin of the peptides as either product-related or HCPs.
Table 5. Analytical and Characterization Electrophoretic Methods for HCP Testing
| Method |
Use |
Advantages |
Disadvantages |
Approximate Sensitivity for HCPs and Ability to Quantify |
|---|---|---|---|---|
| 1-D gel, either reduced or nonreduced, stained with silver or high-sensitivity fluorescent dye. |
Screening a large number of samples for consistency and presence of nonproduct protein bands. May be used as part of GMP control system. |
High resolution separation; provides approximate molecular weight (MW) information; relatively simple to run. Side-by-side analyses of samples provide powerful comparisons for manufacturing consistency. |
Large excess of product protein may obscure HCP bands. Different proteins show differences in staining intensities so only semi-quantitative. Limited capacity of gel lanes for total protein load. |
100 ng/mg. Semiquantitative unless used with an analytical standard for a known HCP. |
| Large-format 2-D gel (e.g., 20 × 25 cm) stained with silver or a high-sensitivity fluorescent dye. |
Screening a small number of samples for consistency and presence of unknown protein spots. Not suitable for routine use in lot release but may be used for characterization of specific lots or HCP standards. |
High-resolution separation of trace HCP impurities from product. Provides approximate MW and pI information on protein spots. |
Large excess of product protein may obscure HCP spots. Different proteins show differences in staining intensities so only semiquantitative. Highly technique-dependent results requiring skilled staff and sophisticated equipment. |
100 ng/mg of total protein load). Semiquantitative unless used with an analytical standard for a known HCP. |
| CE-SDS with laser-induced fluorescence (LIF) detection. Fluorescently labeled samples are separated as either reduced or nonreduced samples. |
Screening a large number of samples for consistency and presence of unknown protein peaks. May be used as part of GMP control system. |
Rapid, high-resolution method to separate fluorescently labeled proteins. |
Sensitivity may not be as high as a 1-D gel with a highly sensitive stain. Fluorescent labels are not specific for HCP proteins, so there is no distinction between HCP and product-related peptides. Difficult to extend to other measures, such as Western blots, or to identify protein peaks. |
1000 ng/mg (0.1% of total protein load). Semiquantitative unless used with an analytical standard for a known HCP. |
5.2 Considerations for Western Blot Methods
The Western blot technique is complementary to the sandwich immunoassay for demonstrating product purity and for characterizing the immunochemical reagents used in the immunoassay (see 3.2.2.2 Characterization of antibodies to HCPs). When the anti-HCP antibody is used as a probe, it may be useful to: 1) detect HCPs that are noncovalently associated with the product and (potentially) missed in immunoassay, or 2) monitor an individual HCP. In other cases, a protein that reacts well in the immunoassay may not be detected in Western blot because a conformational epitope was lost following denaturation. Immunoblots require only one antibody to a given protein, whereas sandwich immunoassays require two. Previous work has shown that high-affinity antibodies that react with a given HCP can be more sensitive than silver or fluorescent dye staining of gels.
1-D Western blots of final product provide a simple reliable orthogonal method for demonstrating product purity. Although the same antibody preparation may be used in both the immunoassay and Western blot, for the reasons noted above, the blot may provide additional information on trace proteins (and miss others), hence its orthogonal nature. Table 6 presents the advantages and disadvantages of 1-D and 2-D Western blot methods for these purposes.
Below is a list of the major considerations for optimization of SDS-PAGE/Western methods:
-
2-D-gel format and size: The larger formats (18-cm length or longer) resolve proteins better and come in a variety of isoelectric focusing (IEF) application modes and molecular weight dimensions (e.g., gradients). Total protein staining of various formats can be studied to optimize the separation in both dimensions and thereby find the balance that separates most (or the maximum number) of HCPs.
-
The load on 2-D gels should be optimized to balance sensitivity needed (higher load) versus resolution desired (lower load). The protein load, ionic content of the sample, and speed of separation need to be balanced to avoid over-heating and to optimize resolution. The stains or substrates to be used will influence this choice. Typically, more is loaded for the immunoblot (e.g., 200 µg) than for the total protein-stained gel or blot (e.g., 50 µg for silver stains). The difference in load may confound the separation to some extent; therefore, finding the right balance will require experimentation.
-
Signal development for both the total protein (gel or blot) and the Western blot requires significant experimentation (e.g., dye types, exposure times, buffer conditions) to optimize. For example, because it is important to detect as many bands as possible, sometimes the signal is amplified so much that the most abundant species obscure the others, or the background increases and contrast is lost. A related issue is standardization and calibration of the densitometer or imager, which must be fine-tuned to achieve consistency under experimentally determined conditions. Both reagent and instrument optimization can require significant time to reproducibly establish the optimized conditions where signal is maximized and background is minimized (see also 1104).
-
The transfer and blocking reagents require experimentation to identify the best conditions (such as buffers, instrument power/time, temperature, and blockers).
Table 6. Analytical and Characterization Western Blot Methods for HCP Testing
| Method |
Use |
Advantages |
Disadvantages |
Approximate Sensitivity and Ability to Quantify HCPs |
|---|---|---|---|---|
| 1-D gel, Western blot |
Screening a large number of samples for consistency and presence of unknown protein bands that react with anti-HCP antibodies. May be used as part of GMP control system. |
High-resolution separation; provides approximate MW information; relatively simple to run. Side-by-side analyses of samples provide powerful comparisons of lots. |
Requires high-quality anti-HCP antibody source. Large excess of product in the gel can lead to nonspecific staining of product bands. SDS-induced denaturation of proteins leads to loss of conformational epitopes. |
Not quantitative for HCPs unless a specific protein standard is used. Sensitivity depends entirely on the quality of the antibody. |
| Large-format 2-D gel (e.g., 20 × 25 cm) Western Blot with chemiluminescence or similar high-sensitivity detection. |
Screening a small number of samples for consistency and presence of unknown protein spots reacting with HCP antibodies. Not suitable for routine use but may be used for characterization of specific lots of product or characterization of antibody reagents. |
High-resolution separation of trace HCP impurities from product. Provides approximate MW and pI information on protein spots. |
Requires high-quality anti-HCP antibody source. Large excess of product protein may obscure HCP spots. SDS-denaturation of proteins leads to loss of conformational epitopes. Produces highly technique-dependent results requiring skilled staff and sophisticated equipment. |
Not quantitative for HCPs unless a specific protein standard is used. Sensitivity depends entirely on the quality of the antibody, but 100 ng/mg is possible. |
5.3 Considerations for Chromatographic and Proteomic Methods
Mass spectrometric techniques for the detection, identification, and quantitation of individual HCPs are rapidly evolving and leverage much of the technology developed for proteomics. Furthermore, as new genomic databases become available (e.g., the CHO genome), mass spectrometric approaches are more useful. These methods (see also Applications of Mass Spectrometry 1736 for additional information) often combine sample preparation such as reduction, alkylation, and proteolytic digestion, followed by separation (e.g., with reversed-phase chromatography RP-HPLC) before introduction into a mass spectrometer that fragments all proteins thus providing an amino acid sequence for each peptide. The resulting sequence information is compared to the product sequence to identify product-related fragments and to a database related to the host (e.g., E. coli, CHO) to identify HCPs.
A challenge for MS analysis stems from the overwhelming number of product-derived peptides relative to impurity peptides. One approach to address this issue of competitive ionization of the peptides (also called ion suppression of the HCP peptides by the product peptides), is to apply LC-MS/MS analysis on partially resolved HCP preparations (e.g., HPLC fractions). This purification reduces the product contribution to total ions in the mass spectrometer. Other approaches are in development. Recent technological improvements such as 1) chromatography resins able to resolve effectively using MS-friendly mobile phases, 2) improved interfaces to front end LC and/or IEF separation systems, and 3) mass spectrometers with higher mass resolution, accuracy, and faster scan rates, now make it possible to identify and quantify specific HCPs in DS with a high degree of confidence. Major challenges in terms of sensitivity and quantitation of sufficiently large sets of heterogeneous HCPs, cost, and QC-related issues remain to be met before this technology can replace immunoassays for the control of HCPs in DS.
As a characterization method orthogonal to the HCP immunoassay, LC-MS/MS data may be used in two ways. First, if the MS-based method does not find HCPs in samples that were also below QL in the immunoassay, then these orthogonal techniques can increase confidence that the HCP ELISA did not miss an HCP that has co-purified. The detection limit for many LC-MS/MS methods is currently in the range of about 10 to 100 ng of HCP per mg of product. It is therefore sufficiently sensitive to rule out a single HCP being present at a high level and is more sensitive than gels. Second, if an impurity is identified at a high level or one that is a safety concern, then one can assess whether the purification process needs to be improved to remove it, whether the current control system is adequate, and whether development of a specific assay is required. Absence of signal does not prove absence of HCPs, but once an HCP is identified, the sensitivity of LC-MS/MS can be enhanced. LC-MS/MS is complementary to other HCP technologies and is increasingly valuable as an orthogonal method. Detailed discussion of all the proteomic approaches to the analysis of HCPs is beyond the scope of this chapter, in part because the field is evolving so rapidly that any review would become out of date quickly.
Intact HCPs also may be separated using HPLC with UV detection. The profile has been used for comparing different lots of null cell runs, for example. Readers are referred to Chromatography 621 for the core science behind HPLC chromatographic methods. Because HCPs differ in abundance, and the chromatography may not provide unique resolution of all proteins, an individual HCP must constitute about 1% (or less with good resolution) of the total protein population for possible detection (even then, a given HCP may be obscured by product, depending on its retention time). Nonetheless, investigation of unknown peaks as a general practice and comparisons of the chromatographic profiles of null cell run HCPs may be useful in determining the similarity of HCPs produced under different culture conditions.
As with the electrophoretic methods discussed in this chapter, reversed-phase separation of proteolytically cleaved product samples with UV detection may already be included in a GMP control system as part of product testing. The presence of new or unexpected peaks in the UV chromatogram may indicate the presence of an HCP impurity, although this method is not overly sensitive (~1% impurity if not obscured by other peaks). However, because no new testing or sample preparation is required, this opportunity to use an orthogonal method for determining purity should be considered.
Table 7. Analytical and Characterization Chromatographic and Proteomic Methods for HCP Testing
| Method |
Use |
Advantages |
Disadvantages |
Approximate Sensitivity and Ability to Quantify |
|---|---|---|---|---|
| Mass Spectrometry – HPLC/electrospray ionization (ESI)-MS/MS or cIEF/ESI-MS/MS or MALDI-TOF of protein digests |
Identification of individual HCPs. May be applied to the characterization of a purified product lot, or null cell run HCP standards. |
Allows identification and monitoring of individual HCPs. May be combined with gels to identify the individual proteins seen in gels. |
Low-throughput method requiring significant sample preparation, skilled staff, and sophisticated instrumentation. |
100 ng/mg and semi-quantitative if used in the discovery mode for unknown HCPs. 10 ng/mg (or better may be possible) and quantitative if used with an analytical standard for a known HCP. |
| RP-HPLC/UV or RP-UHPLC/UV. May be applied to intact proteins or proteolytic digests |
Suitable for comparing null cell run standards if proteins are well separated. Larger proteins may need limited proteolysis or reduction to improve resolution. |
High resolution, separates HCPs based on hydrophobicity and size; direct comparison of null strain and product strain is possible via overlays. |
Difficult to find a stationary phase and mobile phase combination able to resolve and elute all HCPs. Co-elution may hinder quantitation of specific proteins. High concentration of product peak(s) may interfere with detection of trace HCPs. |
10,000 ng/mg (1%). Semi-quantitative unless used with an analytical standard for a known HCP. |
5.4 Concluding Remarks on Supporting Technologies for HCPs
A number of various hybrid analytical techniques have been attempted over the years. One example is fractionation of HCP standards by ion exchange, followed by RP chromatography, and then an immunoassay or MS analysis (2-D MS) on the proteins in the final fractions. Alternatively, “product subtraction” has been evaluated to remove the bulk of the total protein from final product samples before gel electrophoresis of LC-MS/MS analysis. These hybrid methods for coverage determination are very labor intensive, and they may introduce artifacts or biases of their own (e.g., antibodies to product do not typically remove all the myriad of heterogeneous forms). As such, they have not found widespread application, although in individual cases they may provide insight into the nature of product impurities.
Electrophoresis, Western blotting, and immunoassay remain the most widely used methods for HCP reagent characterization and for demonstration of HCP clearance by purification processes, because they are robust, sensitive, and can cover a broad range of protein impurities. Immunoassay and (increasingly) mass spectrometry are highly complementary and the most powerful methods for monitoring residual HCP levels in samples and confirming their absence in final DSs.
6. USE OF HCP IMMUNOASSAYS FOR PROCESS DEVELOPMENT, CHARACTERIZATION, AND VALIDATION
As described previously, although HCP testing has limitations as quantitative assays, if the immunoassay can rapidly and robustly detect most of the HCPs, then it is extremely valuable during process development, characterization, and validation. HCP clearance at each intermediate process step under a variety of conditions is valuable data. Application to process validation requires that the assay be demonstrated to be scientifically sound, suitable for its intended purpose, and appropriately documented. As stated earlier, process pools can differ widely in terms of buffer components, product concentration, and HCP composition. At a minimum, acceptable spike recovery of the HCP standard into in-process pool samples is necessary to qualify test dilutions of the various pools in the assay. In addition, testing each sample type at multiple dilutions is helpful during development to verify that: 1) results are within the range of the assay, and 2) nonlinearity in sample dilution is considered. In cases where the process is shown to clear HCPs robustly, process validation may also be used to justify not having a routine HCP test as part of the cGMP control system.
Figure 5 illustrates data gathered during process development and validation and shows the dilution dependence for a number of in-process pools (Pool 1 is after the first CCF purification step, Pool 2 the next, and so on) through the process. Figure 5 shows HCP ratio (in ng/mg) on the y-axis plotted against the product dilution in the test well on the x-axis. Each sample is serially diluted through three or four twofold dilutions. The starting dilution is determined by both the need to dilute samples with high levels of HCP into the range of the assay and the need to test only at dilutions where acceptable spike recovery has already been demonstrated. In Figure 5, the CCF has slightly more than 1 million ng of HCP per mg of product, meaning that a little less than one-half of the total protein in the CCF is product. The first column pool reduces the HCP level to just over 10,000 ng/mg. The second column has minimal HCP clearance; the third column reduces the HCP ratio to 1,000 ng/mg. The final column in the process reduces the HCP level by an additional 2 logs, resulting in a final HCP ratio of 10 ng/mg. All of the intermediate pools show HCP ratio values that are independent of the dilution of the sample.
+'&img=../../images/v38332/c1132-fig5.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 5. Example of HCP clearance by each purification step. Within a sample set, the first value is the most dilute in its dilution series.
Figure 6 presents a set of data for a different recovery process. As in Figure 5, the HCP ratio data for a serial twofold dilution series of each in-process pool sample are plotted. For CCF and the column 1 and column 2 pools, the HCP ratio (ng/mg) is independent of sample dilution (dilution linearity is achieved as in Figure 5). But after processing over the third column, distinct dilution dependence is apparent in the assay. This nonlinear dilution is likely associated with antigen excess relative to available antibody for the particular set of HCP(s) in the sample (i.e., a single or limited number of HCP species co-purify with the product). With product purification, more product must be put in the assay well for residual HCPs to be detectable in immunoassay. The product concentration in the assay may be increased with each purification step, and thus there is a similar increase in the concentration of the co-purifying HCP impurity. By column 3, the amount of the impurity likely exceeds the capacity of the available antibodies. This phenomenon is repeated in the final pool, which involves no HCP clearance but only buffer exchange by UF/DF. The reported value for these pools is the one obtained by diluting the sample to near the QL of the assay (the most dilute sample in the series) using a method described in Table 4, which is about 300 ng/mg in this case. Additional discussion of assay nonlinearity is found in the 4.3 Sample Linearity subsection of 4. HCP Immunoassay Method Validation.
+'&img=../../images/v38332/c1132-fig6.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 6. Example of HCP clearance with a purification process. Within a sample set, the first value is the most dilute in its dilution series.
6.1 Assays for Individual HCPs
In some cases, a manufacturing process might result in production of relatively high levels of a single HCP that can result in a bias in the reported results. As discussed above, in this situation the concentration of antibodies present in the total HCP assay may not be sufficient to capture all of that particular HCP present. In such cases, where a single, known HCP is present, another assay for that single HCP may be needed. For example, in the case of E. coli production cultures, co-expressing chaperones or other processing enzymes that aid in folding or disulfide formation can be produced in very high levels. In addition, these chaperones may be present in the immunogen used to develop the HCP assay reagents, in a conformation that is different from the way the protein is presented in samples. Thus, antibodies produced to chaperones not bound to product may not recognize, or may poorly recognize, product-bound HCPs. In this case, separate assays using appropriate antibodies may need to be developed. The existence of high levels of a single HCP is not unique to bacterial cell cultures and is possible with any cellular expression system. Another example is when a protein is intentionally added as part of the processing, then it is treated as a single HCP, and a specific assay will provide better data (than the total HCP immunoassay).
6.2 Control Strategy
6.2.1 targets, alert limits, and reject limits
In both United States and European Union regulations, and generally in other markets, HCPs are described as process-related impurities, and guidances state that their presence should be minimized by the use of appropriate, well-controlled manufacturing processes. U.S. 21 CFR 610.13 requires biological products to be “...free of extraneous material except that which is unavoidable...”. ICH Q6B indicates that biologic product manufacturers should assess impurities that may be present and develop acceptance criteria for the impurities based on their preclinical and clinical experience. However, regulatory guidance also recognizes the inherent challenges involved in using assay-specific data and product- and process-specific reagents. Guidance states “The absolute purity of biotechnological and biological products is difficult to determine and the results are method-dependent” (ICH Q6B), and, “In the same manner, standardization of the analytical methods would be problematic since the reagents used are product and production system-related” [European Agency for the Evaluation of Medicinal Products (EMEA) 1997].
Although FDA guidance describes the need to test for HCPs, it also allows, through validation, removal of the release testing requirement, once clearance has been shown to be consistent. An expectation exists that, whenever possible, impurities that were introduced by manufacturing processes and remain in products should be below detectable levels based on a highly sensitive analytical method. However, an appropriately conducted clearance study performed as part of process validation can be an acceptable substitute for lot-to-lot (C of A) testing (FDA 1997), even when low levels of HCPs are present in the DS. In addition, “If impurities are qualitatively and quantitatively (i.e., relative amounts and/or concentrations) the same as in the DS, testing of the DP is not necessary” (ICH Q6B). This is the case for HCPs, the concentration of which in the DP may be inferred from the DS, because the manufacturing process does not introduce, or clear, HCPs when the DS is converted to the DP. Thus, in these instances, HCP testing of the DP is generally not required.
The overarching goal is to develop processes that minimize the quantities of residual HCPs in DSs. When using HCP immunoassays as part of a manufacturing control system, limits for acceptable results need to be established. The “target” for HCP in final product gives the purification development group an objective for the process, which may or may not be achievable. Antibody processes with highly selective affinity chromatography steps (e.g., using Protein A) are sometimes able to achieve a sensitivity of 1 ng of HCP per mg of protein. Target HCP levels determine the design criteria that are then used to develop the process.
Preclinical toxicology lots are the first lots for which official HCP values should be determined, because at this point in product development, the investigator is building the data set that will be used to support the safety of subsequent clinical studies. As the molecule advances through clinical development, the preceding studies are used to support the projected safety (and risk) associated with subsequent studies. Ideally, changes in HCP level and thus clinical exposure should decrease as the product moves through clinical development.
As part of an overall control strategy, the “reject limits” are those levels which, if exceeded, render the product no longer fit for use. During product development, knowledge about the process and the product increases; therefore, it is expected that reject limits can be tightened as one moves from toxicology material to phase I/II material, to phase III, and finally to commercial material. Each drug candidate profile and its clinical program are unique; hence, no single numerical limit is applicable to all.
Reject limits are primarily concerned with patient safety. Typically, one does not have specific data documenting “unsafe” levels of HCPs. Rather, acceptable levels for early stage products should be determined through a risk assessment (including non-clinical data, available literature, previous experience with products manufactured using the same or a similar cell line, etc.). For commercial products, the acceptable levels are also based on the experience gathered during clinical trials. Because the HCP impurity profiles may be influenced by the purification process, the registration batches used in the pivotal clinical trials must be representative of the to-be-marketed process. Reject limits for commercial product should be set to exclude HCP values that are outside the range of clinical experience upon which the risk/benefit decision was made for the biopharmaceutical.
Alert limits, trend limits, or action limits (different companies use different terms) reflect considerations of manufacturing consistency. In a commercial process, there should be sufficient manufacturing history to be able to define out-of-trend results. However, early in product development, where there is little or no manufacturing history, action limits may be used to set upper limits on the expected HCPs, based on experience with similar products for which there is a history, or in comparison to the target value. Exceeding these limits should trigger an investigation to determine whether the lot is releasable. Orthogonal measures of product purity are valuable in such investigations.
With the emergence of orthogonal measures of purity, any non-product related atypical signals should be evaluated. Efforts should be made to revise the purification process to remove any unwanted HCPs present at higher than desirable levels (based on, for example, clinical and non-clinical data, available literature, etc.), or a specific justification of its acceptability must be provided. This is accomplished as part of a risk analysis that considers the clinical exposure similar to that described above for immunoassays.
In cases where HCP assays are improved or changed due to unforeseen events, the reject limits may need to be modified, even increased in some cases (if the new assay has broader coverage). However, it is essential that samples be tested in bridging studies to ensure that no changes in quality would be missed in the new, improved assay. In some cases, improved assays give higher values, so bridging studies (head-to-head with original versus improved assay) will be needed to justify the new specification limit.
6.2.2 safety considerations in setting limits
HCP specification limits in units of ng/mg are typically set using process capabilities and also on the basis of clinical experience (i.e., the levels used in trials where the clinical outcomes are known). Although there are several theoretical safety concerns about HCP impurities, the immunogenicity of the impurity is a primary consideration. An immune response may include formation of antibodies directed against the HCP or directed against the therapeutic protein, where the HCP is believed to have acted as an adjuvant. Readers are referred to 1106 for a more in-depth discussion of factors influencing immunogenicity. When patients receive more than one biotherapeutic protein with their associated impurities, immunogenicity is an even greater concern. Although high-dose products may contain a higher mass of residual HCPs per dose and this should be considered during risk assessments, the potential effect is not necessarily predictable of a clinical outcome. In some cases, very low levels of certain HCPs have been shown to have clinical effects, whereas high levels of other HCPs have had none.
Different limits may be set, depending on whether the product has only one or a very limited number of co-purifying HCPs or it has a multiplicity of HCPs, each present as a small fraction of the total HCP level in the product. This could lead to a separate assay and specification for individual HCP(s) that cannot be cleared from the process and are expected to be present in the DS and may have bioactivity.
The host cell origin can also be a consideration. It is possible that HCPs from human cell lines (e.g., HEK293 cells) may pose less immunogenicity risk than HCPs from more distantly related species because of sequence homology. However, if these proteins are normally intracellular, then they might still be immunogenic and seen as “foreign” by the patient’s immune system. If such antibodies develop, then they could cross-react with endogenous human proteins and neutralize or render ineffective one or more necessary biological systems. As a consequence, arguments can be made on both sides; hence, the risks based on production cell line (microbial versus mammalian) are generally believed to be the same and cannot be practically informative a priori.
In addition to considering immunogenicity, the HCP may be bioactive and a risk. For example, a hamster homolog of a therapeutic protein expressed in CHO cells could be sufficiently similar to the product to co-purify. Alternatively, an anti-human cytokine antibody product expressed in CHO cells could bind to the homologous hamster cytokine, and the complex could co-purify with the antibody product. In both cases, the homologous hamster proteins could be biologically active when administered to patients.
7. SUMMARY AND CONCLUSIONS
Biopharmaceutical products should be as free of residual HCPs as reasonably possible. The industry standard for measuring HCPs is a sandwich immunoassay using polyclonal antibodies directed against the broadest possible spectrum of potential impurity proteins. Because the diversity and proportion of HCPs in the calibration standard is never matched exactly to the HCPs in any sample and due to the broad range of antibodies in the immunochemical reagents, different HCP assays will quantify individual HCPs to a differing extent. Thus, the single numerical value readout of the HCP immunoassay is “immunologically weighted”. Highly immunogenic proteins will overquantify relative to the average population, and weakly immunogenic proteins will underquantify. Some proteins may not induce an immune response in the animal species used to generate antibodies, and the HCP immunoassay will be blind to those proteins. Because of this possibility, orthogonal methods for judging product purity also should be included in the control system. The best strategy is to use the HCP immunoassay in conjunction with other analytical tools.
Finally, one should use caution in extrapolating safe HCP values across products. No single specification limit will suffice. Differences in the biological properties of particular HCPs that co-purify with product, the route of administration, the patient population, and product/HCP dose all contribute to possible factors influencing patient risk. Over the history of recombinant biologic therapeutics, there have been examples where individual co-purifying proteins have been immunogenic, acted as adjuvants to increase the patient immune response to the therapeutic protein, or had biological activities themselves. Although these examples exist, they are rare, and overall there is an excellent safety record for biologics. In spite of the limitations and complexities of HCP assays, careful and diligent development and application of these assays have helped to ensure patient safety for millions of doses. HCP analysis continues to be a complex problem demanding time, resources, and careful thought by biopharmaceutical manufacturers with the goal of ensuring that products consistently meet the standard of having as low an HCP impurity level as can reasonably be achieved.
8. BIBLIOGRAPHY
EMEA. 1997. CPMP position statement on DNA and host cell proteins (HCP) impurities, routine testing versus validation studies. London: European Medicines Agency. CPMP/BWP/382/97.
FDA. 1997. Points to consider in the manufacture and testing of monoclonal antibody products for human use. www.fda.gov/CBER/gdlns/ ptc_mab.pdf. http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/OtherRecommendationsforManufacturers/UCM153182.pdf. Accessed 2013-11-01.
ICH. 1999. Guidance for Industry Q6B Specifications: Test procedures and acceptance criteria for biotechnological/biological products. http://www.ich.org/products/guidelines/quality/article/quality-guidelines.html. Accessed 2013-11-01.
ICH. 2005. Validation of analytical procedures: Text and methodologies. Q2(R1). http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf. Accessed 2014-02-26.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Maura C Kibbey, Ph.D.
Senior Scientific Liaison, Biologics & Biotechnology (301) 230-6309 |
(GCBA2010) General Chapters - Biological Analysis |
USP38–NF33 Supplement : No. 2 Page 7647
Pharmacopeial Forum: Volume No. 40(4)