Fexofenadine Hydrochloride Tablets
DEFINITION
Fexofenadine Hydrochloride Tablets contain NLT 95.0% and NMT 105.0% of the labeled amount of fexofenadine hydrochloride (C32H39NO4·HCl).
IDENTIFICATION
• A. Infrared Absorption  197K
197K
Standard solution:
Transfer 60 mg of USP Fexofenadine Hydrochloride RS to a suitable capped tube and add 10 mL of a mixture of acetonitrile and methanol (10:1).
Sample solution:
Transfer an equivalent to 60 mg of fexofenadine hydrochloride, from a sufficient number of weighed and finely powdered Tablets, to a suitable capped tube, and add 10 mL of a mixture of acetonitrile and methanol (10:1).
Analysis:
Shake or mix the Standard solution and Sample solution on a vortex mixer for 1–2 min to disperse the sample. Allow the solution to stand for 10 min, or centrifuge for 2–3 min. Pass the liquid into a 50-mL beaker using a 0.45-µm polytetrafluoroethlyene syringe filter. Evaporate the solvent until about 0.5 mL remains, using a stream of nitrogen with gentle heating (do not exceed 75 ). Add 5 mL of water and 5 drops of dilute hydrochloric acid, and stir to induce precipitation. Chill in an ice bath for 30 min. Filter the solution through a 10- to 15-µm sintered-glass crucible. Dry the precipitate in an air oven for 1 h at 105 oven for 1 h at 105. Prepare a bromide dispersion from the residue.
). Add 5 mL of water and 5 drops of dilute hydrochloric acid, and stir to induce precipitation. Chill in an ice bath for 30 min. Filter the solution through a 10- to 15-µm sintered-glass crucible. Dry the precipitate in an air oven for 1 h at 105 oven for 1 h at 105. Prepare a bromide dispersion from the residue.
Acceptance criteria:
The IR absorption spectrum of the potassium bromide dispersion of the residue from the sample exhibits maxima only at the same wavelengths as that of a potassium bromide dispersion from the Standard.
• B.
The retention time of the major peak in the Sample solution corresponds to that of the Standard solution, as obtained in the Assay.
ASSAY
• Procedure
Solution A:
Glacial acetic acid and water (17:983). Dilute 100 mL of this solution with water to 1 L.
Solution B:
Dilute 15 mL of a solution containing acetonitrile and triethylamine (1:1) with Solution A to 1 L. Adjust with phosphoric acid to a pH of 5.25.
Diluent:
Acetonitrile and Solution A (3:1)
Mobile phase:
Acetonitrile and Solution B (9:16)
Standard stock solution:
0.25 mg/mL of USP Fexofenadine Hydrochloride RS in Diluent
Standard solution:
0.015 mg/mL from the Standard stock solution in Mobile phase
Sample stock solution:
Transfer a sufficient number of whole Tablets (NLT 10) to a suitable volumetric flask, add Solution A (equivalent to 20% of the total flask volume), and shake by mechanical means at a high speed for 30 min or until the Tablets are fully disintegrated and finely dispersed. Add acetonitrile (equivalent to 80% of the total flask volume), and shake by mechanical means for 60 min. Dilute with Diluent to volume. Pass a portion of this solution through a polytetrafluoroethylene filter having a 0.45-µm or finer pore size, and use the filtrate. Dilute, if necessary, with Diluent to obtain a solution containing an equivalent to 1.2 mg/mL of fexofenadine hydrochloride.
Sample solution:
0.018 mg/mL from the Sample stock solution in Mobile phase
Chromatographic system
Mode:
LC
Detector:
UV 220 nm
Column:
4.6-mm × 25-cm; 5-µm packing L11
Column temperature:
35
Flow rate:
1.5 mL/min
Injection size:
20 µL
System suitability
Sample:
Standard solution
Suitability requirements
Tailing factor:
NMT 2.0
Relative standard deviation:
NMT 2.0%
Analysis
Samples:
Standard solution and Sample solution
Calculate the percentage of C32H39NO4·HCl in the portion of Tablets taken:
Result = (rU/rS) × (CS/CU) × 100
| rU | = | = peak response from the Sample solution |
| rS | = | = peak response from the Standard solution |
| CS | = | = concentration of USP Fexofenadine Hydrochloride RS in the Standard solution (mg/mL) |
| CU | = | = nominal concentration of fexofenadine hydrochloride in the Sample solution (mg/mL) |
Acceptance criteria:
95.0%–105.0%
PERFORMANCE TESTS
• Dissolution 711
Test 1
Medium:
0.001 N hydrochloric acid; 900 mL, deaerated
Apparatus 2:
50 rpm
Time:
10 and 30 min
Determine the percentages of the labeled amount of C32H39NO4·HCl dissolved by using the following method.
Solution A:
1.0 g of monobasic sodium phosphate, 0.5 g of sodium perchlorate, and 0.3 mL of concentrated phosphoric acid in 300 mL of water
Mobile phase:
Acetonitrile and Solution A (7:3)
Standard solution:
USP Fexofenadine Hydrochloride RS in Medium to obtain a solution having a known concentration similar to that expected for the solution under test. [Note—A small amount of methanol, not exceeding 0.5% of the total volume, can be used to dissolve fexofenadine hydrochloride. ]
System suitability solution:
0.44 mg/mL of USP Fexofenadine Related Compound A RS in water. Transfer 1.0 mL of this solution into a vial, and add 40 mL of the Standard solution. [Note—A small amount of acetic acid, not exceeding 5% of the total volume, can be used to dissolve fexofenadine related compound A. ]
Sample solution:
Pass portions of the solution under test through a glass fiber filter having a 0.45-µm pore size.
Chromatographic system
Mode:
LC
Detector:
UV 220 nm
Column:
4.6-mm × 10-cm; packing L1
Flow rate:
1 mL/min
Injection size:
2–3 µg column load of fexofenadine hydrochloride
System suitability
Samples:
Standard solution and System suitability solution
Suitability requirements
Resolution:
NLT 2.0 between fexofenadine and fexofenadine related compound A, System suitability solution
Relative standard deviation:
NMT 2.0%, Standard solution
Analysis
Samples:
Standard solution and Sample solution
Calculate the percentage of C32H39NO4·HCl dissolved in the portion of Tablets taken:
Result = (rU/rS) × (CS/L) × D × V × 100
| rU | = | = peak area from the Sample solution |
| rS | = | = peak area from the Standard solution |
| CS | = | = concentration of USP Fexofenadine Hydrochloride RS in the Standard solution (mg/mL) |
| L | = | = Tablet label claim (mg) |
| D | = | = dilution factor of the Sample solution |
| V | = | = volume of Medium, 900 mL |
Tolerances:
NLT 60% (Q) of the labeled amount of C32H39NO4·HCl is dissolved in 10 min; NLT 80% (Q) of the labeled amount of C32H39NO4·HCl is dissolved in 30 min.
Test 2
If the product complies with this test, the labeling indicates that the product meets USP Dissolution Test 2.
Medium:
0.001 N hydrochloric acid; 900 mL
Apparatus 2:
50 rpm. Use paddles and shafts coated with Teflon.
Time:
30 min
Determine the percentages of the labeled amount of C32H39NO4·HCl dissolved by using the following method.
Solution A:
7 mg/mL of ammonium acetate in water. Adjust with glacial acetic acid to a pH of 4.0 ± 0.05.
Mobile phase:
Acetonitrile and Solution A (2:3)
Standard solution 1:
Transfer 20 mg of USP Fexofenadine Hydrochloride RS to a 100-mL volumetric flask. Add 3.0 mL of methanol, and mix. Dilute with Medium to volume.
Standard solution 2:
Transfer 15.0 mL of Standard solution 1 to a 50-mL volumetric flask. Dilute with Medium to volume.
Standard solution 3:
Transfer 7.5 mL of Standard solution 1 to a 50-mL volumetric flask. Dilute with Medium to volume.
Sample solution:
Pass portions of the solution under test through a suitable filter of 0.45-µm pore size.
Chromatographic system
Mode:
LC
Detector:
UV 259 nm
Column:
4.6-mm × 15-cm; packing L11
Flow rate:
1.5 mL/min
Injection size:
10 µL for Standard solution 1 and 30 µL for Standard solutions 2 and 3
System suitability
Sample:
Any of the Standard solutions
Suitability requirements
Tailing factor:
NMT 2.0
Relative standard deviation:
NMT 2.0%
Analysis
Samples:
Standard solutions 1, 2, and 3 and the Sample solution
Calculate the percentage of C32H39NO4·HCl dissolved in the portion of Tablets taken:
Result = (rU/rS) × (CS/L) × V × 100
| rU | = | = peak area from the Sample solution |
| rS | = | = peak area from the Standard solution |
| CS | = | = concentration of the appropriate Standard solution (mg/mL) |
| V | = | = volume of Medium, 900 mL |
| L | = | = Tablet label claim (mg) |
Tolerances:
NLT 75% (Q) of the labeled amount of C32H39NO4·HCl is dissolved.
Test 3
If the product complies with this test, the labeling indicates that the product meets USP Dissolution Test 3.
Medium:
0.001 N hydrochloric acid; 900 mL for Tablets labeled to contain 30 mg or 60 mg, and 1800 mL for Tablets labeled to contain 180 mg
Apparatus 2:
50 rpm
Time:
45 min
Buffer solution:
6.64 g/L of monobasic sodium phosphate monohydrate and 0.84 g/L of sodium perchlorate monohydrate in water. Add 4 mL/L of triethylamine. Adjust with phosphoric acid to a pH of 2.3 ± 0.1.
Mobile phase:
Buffer solution and acetonitrile (65:35)
Standard stock solution:
0.5 mg/mL of USP Fexofenadine Hydrochloride RS in Mobile phase. This solution is stable for 3.5 months under refrigeration or for 18 days at room temperature.
Standard solution:
Dilute the Standard stock solution with Medium to obtain a final concentration of 0.07 mg/mL of USP Fexofenadine Hydrochloride RS. This solution is stable for 8 days under refrigeration or for 24 h at room temperature.
Sample solution:
Pass a portion of the solution under test through a suitable filter of 0.45-µm pore size.
Chromatographic system
Mode:
LC
Detector:
UV 220 nm
Column:
4.6-mm × 10-cm; 5-µm packing L1
Column temperature:
40
Flow rate:
2.5 mL/min
Injection size:
20 µL
System suitability
Sample:
Standard solution
Suitability requirements
Tailing fator:
NMT 2.0
Column efficiency:
NLT 1000 theroretical plates
Relative standard deviation:
NMT 2.0%
Calculate the percentage of fexofenadine hydrochloride dissolved in the portion of Tablets taken:
Result = (rU/rS) × (CS/L) × V × 100
| rU | = | = peak response from the Sample solution |
| rS | = | = peak response from the Standard solution |
| CS | = | = concentration of the Standard solution (mg/mL) |
| L | = | = Tablet label claim (mg) |
| V | = | = volume of Medium, 900 or 1800 mL |
Tolerances:
NLT 75% (Q) of the labeled amount of fexofenadine hydrochloride is dissolved.
• Uniformity of Dosage Units 905:
Meet the requirements
IMPURITIES
Organic Impurities
• Procedure
Solution A, Solution B, Diluent, Mobile phase, Standard stock solution, Sample stock solution, and Sample solution:
Prepare as directed in the Assay.
Standard solution:
0.015 mg/mL of fexofenadine hydrochloride and 0.0045 mg/mL of fexofenadine related compound A from Quantitative limit solution and the Standard stock solution in Mobile phase
System suitability stock solution:
Dilute 4.0 mL of the Standard stock solution with Mobile phase to 100 mL.
System suitability solution:
Dilute 6.0 mL of the System suitability stock solution with Mobile phase to 100 mL.
Quantitative limit solution:
0.05 mg/mL of USP Fexofenadine Related Compound A RS in Diluent
Chromatographic system
Mode:
LC
Detector:
UV 220 nm
Column:
4.6-mm × 25-cm; 5-µm packing L11
Column temperature:
35
Flow rate:
1.5 mL/min
Injection size:
20 µL
System suitability
Samples:
Standard solution and System suitability solution
[Note—For the relative retention times, see Impurity Table 1. ]
Suitability requirements
Resolution:
NLT 7 between fexofenadine and fexofenadine related compound A, Standard solution
Tailing factor:
NMT 2.0, Standard solution
Relative standard deviation:
NMT 6%, System suitability solution; NMT 2.0% and NMT 3.0% for fexofenadine and fexofenadine related compound A, Standard solution
Analysis
Samples:
Standard solution, Sample stock solution, and Sample solution
Calculate the percentage of fexofenadine related compound A in the portion of Tablets taken:
Result = (rU/rS) × (CS/CU) × 100
| rU | = | = peak area of fexofenadine related compound A in the Sample stock solution |
| rS | = | = peak area of fexofenadine related compound A in the Standard solution |
| CS | = | = concentration of fexofenadine related compound A in the Standard solution (mg/mL) |
| CU | = | = concentration of Fexofenadine hydrochloride in the Sample stock solution |
Calculate the percentage of the decarboxylated degradant [(±)-4-[1-hydroxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]-butyl]-isopropylbenzene] in the portion of Tablets taken:
Result = (rU/rS) × (CS/CU) × (1/F) × 100
| rU | = | = peak area of the decarboxylated degradant in the Sample stock solution |
| rS | = | = peak area of fexofenadine in the Standard solution |
| CS | = | = concentration of USP Fexofenadine Hydrochloride RS in the Standard solution (mg/mL) |
| CU | = | = concentration of fexofenadine hydrochloride in the Sample stock solution |
| F | = | = relative response factor (see Impurity Table 1) |
Calculate the percentage of any other impurities in the portion of Tablets taken:
Result = rU/(F × rS + rT) × 100
| rU | = | = peak area for each individual unknown impurity in the Sample stock solution |
| F | = | = difference in concentration between the Sample stock solution and the Sample solution, 66.7 |
| rS | = | = peak area response for fexofenadine in the Sample solution |
| rT | = | = sum of the peak areas of all unknown impurities in the Sample stock solution |
[Note—Disregard any peak below 0.05%. ]
Acceptance criteria
Individual impurities:
See Impurity Table 1.
Total impurities:
NMT 0.5%
Impurity Table 1
| Name | Relative Retention Time |
Relative Response Factor |
Acceptance Criteria, NMT (%) |
|---|---|---|---|
| Fexofenadine related compound A | 1.6 | — | 0.4 |
| Decarboxylated degradant | 6.7 | 1.1 | 0.15 |
| Fexofenadine | 1.0 | — | — |
| Any individual other impurity | — | 1.0 | 0.2 |
ADDITIONAL REQUIREMENTS
• Packaging and Storage:
Preserve in well-closed containers, and store at controlled room temperature.
• Labeling:
When more than one Dissolution test is given, the labeling states the test used only if Test 1 is not used.
• USP Reference Standards 11
USP Fexofenadine Hydrochloride RS 
') + '&img=../../images/v35300/cas-138452-21-8.gif',600,500);)
USP Fexofenadine Related Compound A RS
Benzeneacetic acid, 4-[1-oxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-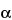 ,-dimethyl.
,-dimethyl.
C32H37NO4 499.65
Benzeneacetic acid, 4-[1-oxy-4-[4-(hydroxydiphenylmethyl)-1-piperidinyl]butyl]-
C32H37NO4 499.65
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| Monograph | Domenick Vicchio, Ph.D.
Senior Scientific Liaison 1-301-998-6828 |
(SM42010) Monographs - Small Molecules 4 |
| Margareth R.C. Marques, Ph.D.
Senior Scientific Liaison 1-301-816-8106 |
(GCDF2010) General Chapters - Dosage Forms | |
| Reference Standards | RS Technical Services 1-301-816-8129 rstech@usp.org |
USP35–NF30 Page 3188
Pharmacopeial Forum: Volume No. 34(4) Page 931