The USP dissolution procedure is a performance test applicable to many dosage forms. It is one test in a series of tests that constitute the dosage form's public specification (tests, procedures for the tests, acceptance criteria). To satisfy the performance test, USP provides the general test chapters Disintegration  701
701 , Dissolution 711, and Drug Release 724. These chapters provide information about conditions of the procedure. For dissolution, these include information about (1) medium, (2) apparatus/agitation rate, (3) study design, (4) assay, and (5) acceptance criteria. Overall the dissolution procedure yields data to allow an accept/reject decision relative to the acceptance criteria, which are frequently based on a regulatory decision. This chapter provides recommendations on how to develop and validate a dissolution procedure.
, Dissolution 711, and Drug Release 724. These chapters provide information about conditions of the procedure. For dissolution, these include information about (1) medium, (2) apparatus/agitation rate, (3) study design, (4) assay, and (5) acceptance criteria. Overall the dissolution procedure yields data to allow an accept/reject decision relative to the acceptance criteria, which are frequently based on a regulatory decision. This chapter provides recommendations on how to develop and validate a dissolution procedure.
GENERAL COMMENTS
The dissolution procedure requires an apparatus, a dissolution medium, and test conditions that provide a method that is discriminating yet sufficiently rugged and reproducible for day-to-day operation and capable of being transferred between laboratories.
The acceptance criteria should be representative of multiple batches with the same nominal composition and manufacturing process, typically including key batches used in pivotal studies, and representative of performance in stability studies.
The procedure should be appropriately discriminating, capable of distinguishing significant changes in a composition or manufacturing process that might be expected to affect in vivo performance. It is also possible for the procedure to show differences between batches when no significant difference is observed in vivo. This situation requires careful evaluation of whether the procedure is too sensitive or appropriately discriminating. Assessing the results from multiple batches that represent typical variability in composition and manufacturing parameters may assist in this evaluation. It is sometimes valuable to intentionally vary manufacturing parameters, such as lubrication, blend time, compression force, or drying parameters, to further characterize the discriminatory power of the procedure.
With regard to stability, the dissolution test should appropriately reflect relevant changes in the drug product over time that are caused by temperature, humidity, photosensitivity, and other stresses.
A properly designed test should result in data that are not highly variable and should not be associated with significant analytical solution stability problems. High variability in results can make it difficult to identify trends or effects of formulation changes. Dissolution results may be considered highly variable if the relative standard deviation (RSD) is greater than 20% at time points of 10 minutes or less and greater than 10% RSD at later time points.1 However, most dissolution results exhibit less variability than this. The source of the variability should be investigated when practical, and attempts should be made to reduce variability whenever possible. The two most likely causes are the formulation itself (e.g., drug substance, excipients, or manufacturing process) or artifacts associated with the test procedure (e.g., coning, tablets sticking to the vessel wall or basket screen). Visual observations are often helpful for understanding the source of the variability and whether the dissolution test itself is contributing to the variability. Any time the dosage contents do not disperse freely throughout the vessel in a uniform fashion, aberrant results can occur. Depending on the problem, the usual remedies include changing the apparatus type, speed of agitation, or deaeration; consideration and/or examination of sinker type; and changing the composition of the medium. Modifications to the apparatus may also be useful, with proper justification and validation.
Many causes of variability can be found in the formulation and manufacturing process. For example, poor content uniformity, process inconsistencies, a reaction taking place at different rates during dissolution, excipient interactions or interference, film coating, capsule shell aging, and hardening or softening of the dosage form on stability may be sources of variability and interferences. During routine testing of the product, variability outside the expected range should be investigated from analytical, formulation, and processing perspectives.
MEDIUM
Physical and chemical data for the drug substance and dosage unit need to be determined before selecting the dissolution medium. Two key properties of the drug are the solubility and solution state stability of the drug as a function of the pH value. When selecting the composition of the medium, the influence of buffers, pH value, and surfactants on the solubility and stability of the drug need to be evaluated. Key properties of the dosage unit that may affect dissolution include release mechanism (immediate, delayed, or modified) and disintegration rate as affected by hardness, friability, presence of solubility enhancers, and presence of other excipients.
Generally, when developing a dissolution procedure, one goal is to have sink conditions, defined as the volume of medium at least three times that required in order to form a saturated solution of drug substance. When sink conditions are present, it is more likely that dissolution results will reflect the properties of the dosage form. A medium that fails to provide sink conditions may be acceptable if it is shown to be more discriminating or otherwise appropriately justified.
Using an aqueous–organic solvent mixture as a dissolution medium is discouraged; however, with proper justification this type of medium may be acceptable.
Purified water is often used as the dissolution medium, but is not ideal for several reasons. First, the quality of the water can vary depending on the source of the water, and the pH value of the water is not controlled. Second, the pH value can vary from day to day and can also change during the run, depending on the active substance and excipients. Despite these limitations, water is inexpensive, readily available, easily disposed of, ecologically acceptable, and suitable for products with a release rate independent of the pH value of the medium.
The dissolution characteristics of an oral formulation should be evaluated in the physiologic pH range of 1.2 to 6.8 (1.2 to 7.5 for modified-release formulations). During method development, it may be useful to measure the pH before and after a run to discover whether the pH changes during the test. Selection of the most appropriate conditions for routine testing is then based on discriminatory capability, ruggedness, stability of the analyte in the test medium, and relevance to in vivo performance, where possible.
Typical media for dissolution may include the following (not listed in order of preference): dilute hydrochloric acid, buffers in the physiologic pH range of 1.2 to 7.5, simulated gastric or intestinal fluid (with or without enzymes), water, and surfactants (with or without acids or buffers) such as polysorbate 80, sodium lauryl sulfate, and bile salts.
The molarity of the buffers and acids used can influence the solubilizing effect, and this factor may be evaluated.
For compounds with high solubility and high permeability (as defined by the Biopharmaceutics Classification System), the choice of medium and apparatus may be influenced by the referenced FDA Guidance1.
For very poorly soluble compounds, aqueous solutions may contain a percentage of a surfactant (e.g., sodium lauryl sulfate, polysorbate, or lauryldimethylamine oxide) that is used to enhance drug solubility. The need for surfactants and the concentrations used can be justified by showing profiles at several different concentrations. Surfactants can be used either as wetting agents or to solubilize the drug substance.
Volume
Normally, for basket and paddle apparatus, the volume of the dissolution medium is 500 mL to 1000 mL, with 900 mL as the most common volume. The volume can be raised to between 2 and 4 L, using larger vessels and depending on the concentration and sink conditions of the drug; justification for this procedure is expected.
Deaeration
The significance of deaeration of the medium should be determined, because air bubbles can interfere with the test results, acting as a barrier to dissolution if present on the dosage unit or basket mesh. Further, bubbles can cause particles to cling to the apparatus and vessel walls. On the other hand, bubbles on the dosage unit may increase buoyancy, leading to an increase in the dissolution rate, or may decrease the available surface area, leading to a decrease in the dissolution rate. A dearation method is described as a footnote in the Procedure section under Dissolution 711. Typical steps include heating the medium, filtering, and drawing a vacuum for a short period of time. Other methods of deaeration are available and in routine use throughout the industry. Media containing surfactants are not usually deaerated because the process results in excessive foaming. To determine whether deaeration of the medium is necessary, results from dissolution samples run in nondeaerated medium and deaerated medium should be compared.
Enzymes
The use of enzymes in the dissolution medium is permitted in accordance with Dissolution 711 when dissolution failures occur as a result of cross-linking with gelatin capsules or gelatin-coated products.
In Vitro–In Vivo Correlation (IVIVC)
An in-depth discussion on IVIVC can be found in In Vitro and In Vivo Evaluation of Dosage Forms 1088. A brief discussion follows.
Biorelevant medium is a medium that has some relevance to the in vivo performance of the dosage unit. Choice of a biorelevant medium is based on (1) a mechanistic approach that considers the absorption site, if known, and (2) whether the rate-limiting step to absorption is the dissolution or permeability of the compound. In some cases, the biorelevant medium will be different from the test conditions chosen for the regulatory test, and the time points are also likely to be different. If the compound dissolves quickly in the stomach and is highly permeable, gastric emptying time may be the rate-limiting step to absorption. In this case, the dissolution test should demonstrate that the drug is released quickly under typical gastric (acidic) conditions. On the other hand, if dissolution occurs primarily in the intestinal tract (e.g., for a poorly soluble, weak acid), a higher pH range (e.g., simulated intestinal fluid with a pH of 6.8) may be more appropriate. The fed and fasted states may also have significant effects on the absorption or solubility of a compound. Compositions of media that simulate the fed and fasted states can be found in the literature. These media reflect changes in pH, bile concentrations, and osmolarity after meal intake and therefore have a composition different from that of typical compendial media. They are primarily used to establish in vitro–in vivo correlations during formulation development and to assess potential food effects and are not intended for quality control purposes. For quality control purposes, the substitution of natural surfactants (bile components) with appropriate synthetic surfactants is permitted and encouraged because of the expense of the natural substances and the labor-intensive preparation of the biorelevant media.
APPARATUS/AGITATION
Apparatus
The choice of apparatus is based on knowledge of the formulation design and the practical aspects of dosage form performance in the in vitro test system. For solid oral dosage forms, Apparatus 1 and Apparatus 2 are used most frequently.
When Apparatus 1 or 2 is not appropriate, another official apparatus may be used. Apparatus 3 (Reciprocating Cylinder) has been found to be especially useful for bead-type modified-release dosage forms. Apparatus 4 (Flow-Through Cell) may offer advantages for modified-release dosage forms that contain active ingredients with limited solubility. In addition, Apparatus 3 or Apparatus 4 may have utility for soft gelatin capsules, bead products, suppositories, or poorly soluble drugs. Apparatus 5 (Paddle over Disk) and Apparatus 6 (Rotating Cylinder) have been shown to be useful for evaluating and testing transdermal dosage forms. Apparatus 7 (Reciprocating Holder) has been shown to have application to nondisintegrating oral modified-release dosage forms, as well as to transdermal dosage forms.
Some changes can be made to the apparatus; for example, a basket mesh size other than the typical 40-mesh basket (e.g., 10, 20, 80 mesh) may be used when the need is clearly documented by supporting data. In countries where available mesh sizes vary from the USP-specified mesh value, basket material with the nearest metric dimension should be used. Care must be taken that baskets are uniform and meet the dimensional requirements specified under Dissolution 711. If the basket screens become clogged during dissolution of capsule or tablet formulations, it may be advisable to switch to the paddle method. The volume can be increased from the typical 900 to 1000 mL by using 2- and 4-L vessels to assist in meeting sink conditions for poorly soluble drugs.
A noncompendial apparatus may have some utility with proper justification, qualification, and documentation of superiority over the standard equipment. For example, a small-volume apparatus with mini paddles and baskets may be considered for low-dosage strength products. The rotating bottle or static tubes (jacketed stationary tubes enclosed with a water jacket and equipped with a magnetic stirrer) may also have utility for microspheres and implants, peak vessels for eliminating coning, and modified flow-through cells for special dosage forms, including powders and stents.
Sinkers
When sinkers are used, a detailed description of the sinker must be stated in the written procedure. It may be useful to evaluate different sinkers, recognizing that sinkers can significantly influence the dissolution profile of a dosage unit. When transferring the procedure, the sinkers should be duplicated as closely as possible in the next facility. There are several types of commercially available sinkers. A method for making sinkers by hand, sinkers that are similar to “a few turns of wire helix” as described in Apparatus 2 (Paddle Apparatus) under Dissolution 711, is described below.
Materials—
Use 316 stainless steel wire or other inert material, typically 0.032 inch/20 gauge; and cylinders of appropriate diameter (e.g., cork borers). Sizes are shown in the accompanying table.
| Capsule Shell Type |
Length of Wire (cm) |
Diameter Size (cm) |
Cork Bore Number |
|---|---|---|---|
| #0, elongated | 12 | 0.8 | 4 |
| #1 and #2 | 10 | 0.7 | 3 |
| #3 and #4 | 8 | 0.55 | 2 |
Procedure—
Cut the specified length of wire, coil around a cylinder of the appropriate size, and use small pliers to curve in the ends. Use caution, because wire ends may be rough and may need to be filed.
If the sinker is handmade, the sinker material and construction procedure instructions should be documented; if a commercial sinker is used, the vendor part number should be reported.
Agitation
For immediate-release capsule or tablet formulations, Apparatus 1 (baskets) at 100 rpm or Apparatus 2 (paddles) at 50 or 75 rpm are most commonly used. Other agitation speeds and apparatus are acceptable with appropriate justification.
Rates outside 25 to 150 rpm are usually inappropriate because of the inconsistency of hydrodynamics below 25 rpm and because of turbulence above 150 rpm. Agitation rates between 25 and 50 rpm are generally acceptable for suspensions. For dosage forms that exhibit coning (mounding) under the paddle at 50 rpm, the coning can be reduced by increasing the paddle speed to 75 rpm, thus reducing the artifact and improving the data. If justified, 100 rpm may be used, especially for extended-release products. Decreasing or increasing the apparatus rotation speed may be justified if the profiles better reflect in vivo performance and/or the method results in better discrimination without adversely affecting method reproducibility.
Selection of the agitation and other study design elements for modified-release dosage forms is similar to that for immediate-release products. These elements should conform to the requirements and specifications given in Dissolution 711 when the apparatus has been appropriately calibrated.
STUDY DESIGN
Time Points
For immediate-release dosage forms, the duration of the procedure is typically 30 to 60 minutes; in most cases, a single time point specification is adequate for Pharmacopeial purposes. Industrial and regulatory concepts of product comparability and performance may require additional time points, which may also be required for product registration or approval. A sufficient number of time points should be selected to adequately characterize the ascending and plateau phases of the dissolution curve. According to the Biopharmaceutics Classification System referred to in several FDA Guidances, highly soluble, highly permeable drugs formulated with rapidly dissolving products need not be subjected to a profile comparison if they can be shown to release 85% or more of the active drug substance within 15 minutes. For these types of products a one-point test will suffice. However, most products do not fall into this category. Dissolution profiles of immediate-release products typically show a gradual increase reaching 85% to 100% at about 30 to 45 minutes. Thus, dissolution time points in the range of 15, 20, 30, 45, and 60 minutes are usual for most immediate-release products. For rapidly dissolving products, including suspensions, useful information may be obtained from earlier points, e.g., 5 to 10 minutes. For slower-dissolving products, time points later than 60 minutes may be useful. Dissolution test times for compendial tests are usually established on the basis of an evaluation of the dissolution profile data.
So-called infinity points can be useful during development studies. To obtain an infinity point, the paddle or basket speed is increased at the end of the run for a sustained period (typically 15 to 60 minutes), after which time an additional sample is taken. Although there is no requirement for 100% dissolution in the profile, the infinity point can provide data that may supplement content uniformity data and may provide useful information about formulation characteristics during initial development or about method bias.
For an extended-release dosage form, at least three test time points are chosen to characterize the in vitro drug release profile for Pharmacopeial purposes. Additional sampling times may be required for drug approval purposes. An early time point, usually 1 to 2 hours, is chosen to show that there is little probability of dose dumping. An intermediate time point is chosen to define the in vitro release profile of the dosage form, and a final time point is chosen to show the essentially complete release of the drug. Test times and specifications are usually established on the basis of an evaluation of drug release profile data. For products containing more than a single active ingredient, drug release is to be determined for each active ingredient.
Observations
Visual observations and recordings of product dissolution and disintegration behavior are very useful because dissolution and disintegration patterns can be indicative of variables in the formulation or manufacturing process. To accomplish visual observation, proper lighting (with appropriate consideration of photodegradation) of the vessel contents and clear visibility in the bath are essential. Documenting observations by drawing sketches and taking photographs or videos can be instructive and helpful for those who are not able to observe the real time dissolution test. Observations are especially useful during method development and formulation optimization. Examples of typical observations include, but are not limited to, the following:
- Uneven distribution of particles throughout the vessel. This can occur when particles cling to the sides of the vessel, when there is coning or mounding directly under the apparatus, when particles float at the surface of the medium, when film-coated tablets stick to the vessel, and/or when off-center mounds are formed.
- Air bubbles on the inside of the vessel or on the apparatus or dosage unit. Sheen on the apparatus is also a sign of air bubbles. This observation would typically be made when assessing the need to deaerate the medium.
- Dancing or spinning of the dosage unit, or the dosage unit being hit by the paddle.
- Adhesion of particles to the paddle or the inside of the basket, which may be observed upon removal of the stirring device at the end of the run.
- Pellicles or analogous formations, such as transparent sacs or rubbery, swollen masses surrounding the capsule contents.
- Presence of large floating particles or chunks of the dosage unit.
- Observation of the disintegration rate (e.g., percentage reduction in size of the dosage unit within a certain time frame).
- Complex disintegration of the coating of modified or enteric-coated products—for example, the partial opening and splitting apart (like a clamshell) or incomplete opening of the shell accompanied by the release of air bubbles and excipients.
Sampling
Manual—
Manual sampling uses plastic or glass syringes, a stainless steel cannula that is usually curved to allow for vessel sampling, a filter, and/or a filter holder. The sampling site must conform to specifications under Dissolution 711.
Autosampling—
Autosampling is a useful alternative to manual sampling, especially if the test includes several time points. However, because regulatory labs may perform the dissolution test using manual sampling, autosampling requires validation with manual sampling.
There are many brands of autosamplers, including semiautomated and fully automated systems. Routine performance checks, cleaning, and maintenance as described in the pertinent standard operating procedures or metrology documents are useful for reliable operation of these devices.
Some instruments are equipped with sampling through the basket or paddle shaft. Proper validation (e.g., demonstrated equivalence to results with the usual sampling procedure) may be required.
The disturbance of the hydrodynamics of the vessel by sampling probes should be considered and adequate validation performed to ensure that the probes are not introducing a significant change in the dissolution rate.
Comparison of manual and automated procedures should be performed to evaluate the interchangeability of the procedures. This can be accomplished by comparing data from separate runs or, in some cases, by sampling both ways from the same vessel. Results should be consistent with the requirements for intermediate precision (described in this chapter in Validation) if the procedures are to be considered interchangeable.
Other aspects of automation validation may include carryover of residual drug, effect of an in-residence probe (simultaneous sampling as mentioned above may not be suitable in this case), adsorption of drug, and cleaning and/or rinse cycles.
Filters
Filtration of the dissolution samples is usually necessary to prevent undissolved drug particles from entering the analytical sample and further dissolving. Also, filtration removes insoluble excipients that may otherwise cause high background or turbidity. Prewetting of the filter with the medium may be necessary.
Filters can be in-line or at the end of the sampling probe or both. The pore size can range from 0.45 to 70 µm. The usual types of filters are depth, disk, and flow-through. However, if the excipient interference is high, if the filtrate has a cloudy appearance, or if the filter becomes clogged, an alternative type of filter or pore size should be evaluated.
Adsorption of the drug(s) onto the filter needs to be evaluated. If drug adsorption occurs, the amount of initial filtrate discarded may need to be increased. If results are still unsuitable, an alternative filter material may be sought.
Filter validation may be accomplished by preparing a suitable standard solution or a completely dissolved sample solution (e.g., prepared as a typical sample in a vessel or a sample put in a beaker and stirred with a magnetic stirrer for 1 hour). For standard solutions, compare the results for filtered solutions (after discarding the appropriate volume) to those for the unfiltered solutions. For sample solutions, compare the results for filtered solutions (after discarding the appropriate volume) to those for centrifuged, unfiltered solutions.
Centrifugation
Centrifugation of samples is not preferred, because dissolution can continue to occur and because there may be a concentration gradient in the supernatant. A possible exception might be for compounds that adsorb onto all common filters.
ASSAY
The usual assay for a dissolution sample is either spectrophotometric determination or HPLC. The preferred method of analysis is spectrophotometric determination because results can be obtained faster, the analysis is simpler, and fewer solvents are used. HPLC methods are used when there is significant interference from excipients or among drugs in the formulation to improve analytical sensitivity and/or when the analysis can be automated. It may be useful to obtain data for the drug with a stability-indicating assay (e.g., HPLC chromatograms) in the medium of choice, even if the primary assay is based on a spectrophotometric method.
VALIDATION
The validation topics described in this section are typical but not all-inclusive. The validation elements addressed may vary, depending on the phase of development or the intended use for the data.2 The acceptance criteria are presented as guidelines only and may differ for some products. Firms should document the appropriate acceptance criteria for their products in pertinent SOPs. Other considerations may be important for special dosage forms. The extent of validation depends on the phase of the product development. Full validation takes place by the time of Phase III clinical studies. Validation studies should address the variations associated with different profile time points. For products containing more than a single active ingredient, the dissolution method needs to be validated for each active ingredient.
Specificity/Placebo Interference
It is necessary to demonstrate that the results are not unduly affected by placebo constituents, other active drugs, or degradates.
The placebo consists of all the excipients and coatings (inks, sinker, and capsule shell are also included when appropriate) without the active ingredient. Placebo interference may be determined by weighing samples of the placebo blend and dissolving or dispersing them in dissolution medium at concentrations that would be encountered during testing. It may be desirable to perform this experiment at 37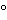 by comparing it to the 100% standard by the formula:
by comparing it to the 100% standard by the formula:
100C(AP/AS)(V/L)
in which C is the concentration, in mg per mL, of the standard; AP and AS are the absorbances of the placebo and the standard, respectively; V is the volume, in mL, of the medium; and L is the label claim, in mg. The interference should not exceed 2%.
Note—For extended-release products, a placebo version of the finished dosage form may be more appropriate to use than blends, because this placebo formulation will release the various excipients in a manner more nearly reflecting the product than will a simple blend of the excipients. In this case, it may be appropriate to evaluate potential interference at multiple sampling points in the release profile.
If the placebo interference exceeds 2%, then method modification—such as (1) choosing another wavelength, (2) baseline subtraction using a longer wavelength, or (3) using HPLC—may be necessary in order to avoid the interference. When other active drugs or significant levels of degradates are present, it is necessary to demonstrate that these do not significantly affect the results. One procedure for doing this is to measure the matrix in the presence and absence of the other active drug or degradate: any interference should not exceed 2%.
Linearity and Range
Linearity and range are typically established by preparing solutions of the drug, ranging in concentration from below the lowest expected concentration to above the highest concentration during release. This may be done in conjunction with accuracy/recovery determination. The scheme may be altered if different flow-cell sizes or injection volumes are used.
Typically, solutions are made from a common stock if possible. For the highest concentration, the determination may not exceed the linearity limits of the instrument.
Organic solvents may be used to enhance drug solubility for the preparation of the standard solutions; however, no more than 5% (v/v) of organic solvent in the final solution should be used, unless validated.
Linearity is typically calculated by using an appropriate least-squares regression program. Typically, a square of the correlation coefficient (r2 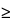 0.98) demonstrates linearity. In addition, the y-intercept must not be significantly different from zero.
0.98) demonstrates linearity. In addition, the y-intercept must not be significantly different from zero.
Accuracy/Recovery
Accuracy/recovery are typically established by preparing multiple samples containing the drug and any other constituents present in the dosage form (e.g., excipients, coating materials, capsule shell) ranging in concentration from below the lowest expected concentration to above the highest concentration during release.
In cases of poor drug solubility, it may be appropriate to prepare a stock solution by dissolving the drug substance in a small amount of organic solvent (typically not exceeding 5%) and diluting to the final concentration with dissolution medium. An amount of stock solution equivalent to the targeted label claim may be added to the vessel instead of the drug powder. Similarly, for very low strengths, it may be more appropriate to prepare a stock solution than to attempt to weigh very small amounts. The measured recovery is typically 95% to 105% of the amount added. Bracketing or matrixing of multiple strengths may be useful.
A special case for validation is the Acid Stage procedure described in Delayed-Release Dosage Forms under Dissolution 711. The limit of not more than 10% needs to be validated. If the compound degrades in acid, the validation experiment must address this fact.
Precision
Repeatability—
Repeatability is determined by replicate measurements of standard and/or sample solutions. It can be measured by calculating the RSD of the multiple injections or spectrophotometric readings for each standard solution, or from the accuracy or linearity data.
Intermediate Precision—
Intermediate precision may be evaluated to determine the effects of random events on the precision of the analytical procedure. This evaluation is typically done later in the development of the drug product. The precision can be across the range of product strengths. Typical variations to study include days, analysts, and equipment. The use of an experimental matrix design is encouraged for evaluation of intermediate precision. If possible, intermediate precision can be evaluated using a well-characterized lot of drug product of tight content uniformity. In cases where a well-characterized product is not available, placebo and active ingredient may be used to identify intermediate precision.
The dissolution profiles on the same sample may be run by at least two different analysts, each analyst preparing the standard solutions and the medium. Typically, the analysts use different dissolution baths, spectrophotometers or HPLC equipment (including columns), and autosamplers; and they perform the test on different days. This procedure may not need to be performed for each strength; instead, bracketing with high and low strengths may be acceptable.
A typical acceptance criterion is that the difference in the mean value between the dissolution results at any two conditions using the same strength does not exceed an absolute 10% at time points with less than 85% dissolved and does not exceed 5% for time points above 85%. Acceptance criteria may be product-specific, and other statistical tests and limits may be used.
Robustness
The evaluation of robustness, which assesses the effect of making small, deliberate changes to the dissolution conditions, typically is done later in the development of the drug product. The number of replicates (typically 3 or 6) is dependent on the intermediate precision.
Parameters to be varied are dependent on the dissolution procedure and analysis type. They may include medium composition (e.g., buffer or surfactant concentration), pH, volume, agitation rate, and temperature. For HPLC analysis, parameters may include mobile phase composition (percentage organic, buffer concentration, pH), flow rate, wavelength, column temperature, and multiple columns (of the same type). For spectrophotometric analysis, the wavelength may be varied.
Standard and Sample Solution Stability
The standard solution is stored under conditions that ensure stability. The stability of the standard is analyzed over a specified period of time, using a freshly prepared standard solution at each time interval for comparison. The acceptable range for standard solution stability is typically between 98% and 102%.
The sample solution is typically stored at room temperature. The sample is analyzed over a specified period of time using the original sample solution response for comparison. The typical acceptable range for sample solution stability may be between 98% and 102% compared with the initial analysis of the sample solutions. If the solution is not stable, aspects to consider could be temperature (refrigeration may be needed), light protection, and container material (plastic or glass).
The procedure may state that the standards and samples need to be analyzed within a time period demonstrating acceptable standard and sample solution stability.
Spectrophotometric Analysis
Samples may be automatically introduced into the spectrophotometer using autosippers and flow cells. Routine performance checks, cleaning, and maintenance as described in the standard operating procedures or metrology documents are useful for reliable operation of these instruments. Cells with path lengths ranging from 0.02 to 1 cm are typically used. Cell alignment and air bubbles could be sources of error. The smaller path length cells are used to avoid diluting the sample; however, acceptable linearity and standard error need to be demonstrated.
During analysis, standard solutions are typically prepared and analyzed at just one concentration at 100% (or the selected Q value) of the dosage strength. During profile analysis, other concentrations may be useful. A typical blank, standard, and sample may be analyzed in a sequence that brackets the sample with standards and blanks, especially at the beginning and end of the analysis.
In most cases, the mean absorbance of the dissolution medium blank may not exceed 1% of the standard. Values higher than 1% must be evaluated on a case-by-case basis. The typical RSD for UV analysis is usually not more than 2%.
The absorptivity is calculated by dividing the mean standard absorbance by the concentration, in mg per mL, divided by the flow-cell path length in cm. After enough historical data are accumulated, an acceptable absorptivity range for the analyte (using the appropriate flow cell) may be determined. This value may be useful in troubleshooting aberrant data.
Fiber optics as a sampling and determinative method, with proper validation, is an option.
It may be useful to examine the UV spectrum of the drug in solution to select the optimum wavelength.
HPLC
For HPLC analysis, the compatibility of dissolution media and mobile phase may be examined, especially if large injector volumes (over 100 µL) are needed. Samples are normally analyzed with HPLC using a spectrophotometric detector and an auto-injector. Single injections of each vessel time point with standards throughout the run constitute a typical run design. System suitability tests include, at a minimum, the retention window and injection precision. Typically, the repeatability of an HPLC analysis should be less than or equal to 2% RSD for five or six standard determinations. The standard level is typically at the 100% label claim level, especially for a single-point analysis.
Preparation of the placebo samples for the HPLC analysis is to be performed in the same way as in the spectrophotometric analysis. Examine the chromatogram for peaks eluting at the same retention time as the drug. If there are extraneous peaks, inject the standard solution, and compare retention times. If the retention times are too close, spike the placebo solution with the drug. Chromatograms may also be obtained over an extended run time using the blank (dissolution medium), standard, and sample solution to identify late eluters that may interfere with subsequent analyses.
The validation documentation may include overlaid representative chromatograms or spectra of blank dissolution medium, a filtered placebo solution, a standard solution, and a filtered dissolution sample. Absence of interfering peaks in the placebo chromatogram or lack of absorbance by the placebo at the analytical wavelength demonstrates specificity.
ACCEPTANCE CRITERIA
Typical acceptance criteria for the amount of active ingredient dissolved, expressed as a percentage of the labeled content (Q), are in the range of 75% to 80% dissolved. A Q value in excess of 80% is not generally used, because allowance needs to be made for assay and content uniformity ranges.3 Acceptance criteria including test times are usually established on the basis of an evaluation of the dissolution profile data. Acceptance criteria should be consistent with historical data, and there is an expectation that acceptable batches (e.g., no significant differences in in vivo performance, composition, or manufacturing procedure) will have results that fall within the acceptance criteria.
1
The Biopharmaceutics Classification System is outlined in the FDA Guidance for Industry: Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System, August 2000; http://www.fda.gov/cder/guidance/3618fnl.htm, accessed 6/22/2005.
2
Boudreau, S.P.; McElvain, J.S.; Martin, L.D.; Dowling, T.; Fields, S.M. Method Validation by Phase of Development, an Acceptable Analytical Practice. Pharmaceutical Technology 2004; 28(11):54–66.
3
See the FDA Guidance for Industry: Dissolution Testing of Immediate-Release Solid Oral Dosage Forms, August 1997; http://www.fda.gov/cder/guidance/1713bp1.pdf, accessed 6/22/2005.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | William E. Brown
Senior Scientific Liaison 1-301-816-8380 |
(GCDF2010) General Chapters - Dosage Forms |
USP35–NF30 Page 675
Pharmacopeial Forum: Volume No. 31(5) Page 1463