The Pharmacopeia provides for dissolution and drug release testing in the majority of monographs for solid oral and transdermal dosage forms. In recognition of the sensitivity of dissolution tests, where a valid bioavailability-bioequivalence (BA-BE) study is in hand, the policy of this Pharmacopeia has been to give this information dominant consideration in setting dissolution standards. Early practice was to develop dissolution requirements based on the in vitro performance of clinically successful formulations. Similarity in dissolution behavior has long been sought from the perspectives of both bioavailability and quality control considerations.
It is the goal of the pharmaceutical scientist to find a relationship between an in vitro characteristic of a dosage form and its in vivo performance. The earliest achievable in vitro characteristic thought to portend an acceptable in vivo performance was tablet and capsule disintegration. A test for disintegration was adopted in USP XIV (1950). At that time, no quantitative work was done in attempting to demonstrate such a relationship, especially in regard to in vivo product performance. However, advances in instrumental methods of analysis ultimately opened up prospects for this work. The disintegration test was recognized as being insufficiently sensitive by the USP-NF Joint Panel on Physiologic Availability, and in 1968 the Panel directed the identification of candidate articles for the first twelve official dissolution tests that used Apparatus 1.
The state of science is such that conduct of in vivo testing is necessary in the development and evaluation of dosage forms. Also, no product, including suspensions and chewable tablets, should be developed without dissolution or drug release characterization where a solid phase exists. This chapter sets forth, for products intended for human use, guidelines for characterizing a drug that include: (1) developing in vitro test methods for immediate-release and modified-release dosage forms, (2) designing in vivo protocols, and (3) demonstrating and assessing in vitro-in vivo correlations for modified-release dosage forms.
IN VITRO EVALUATION
Dissolution and Drug Release Testing—Method Development for Immediate-Release Dosage Forms
Dissolution testing is required for all solid oral Pharmacopeial dosage forms in which absorption of the drug is necessary for the product to exert the desired therapeutic effect. Exceptions are for tablets meeting a requirement for completeness of solution or for rapid (10 to 15 minutes) disintegration for soluble or radiolabeled drugs. The apparatus and procedure conform to the requirements and specifications given in the general chapter Dissolution  711
711 . Generally, experiments are conducted at 37
. Generally, experiments are conducted at 37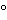 .
.
The dissolution medium preferably is deaerated water or, if substantiated by the solubility characteristics of the drug or the formulation, a buffered aqueous solution (typically pH 4 to 8) or a dilute acid (0.001 N to 0.1 N hydrochloric acid) may be used. The usual volume of the medium is 500 to 1000 mL, with the use of greater volumes (up to 2000 mL) allowed for drugs having limited solubility. The quantity of medium used should be not less than 3 times that required to form a saturated solution of the drug substance. The significance of deaeration of the medium should be determined. Addition of solutes (i.e., surfactants) and electrolytes to aid in solubilization of the drug must be balanced against the loss of the discriminatory power of the test. The use of hydroalcoholic media is generally not favored. The use of such media should be supported by a documented in vitro-in vivo correlation. Conversely, it should be recognized that this discriminatory power could in some circumstances be excessive in that it may result in detection of differences in dissolution that are not clinically significant.
The choice of apparatus should be based on knowledge of the formulation design and actual dosage form performance in the in vitro test system. Since dissolution apparatuses tend to become less discriminating when operated at faster speeds, lower stirring speeds should be evaluated and an appropriate speed chosen in accordance with the test data. The most common operating speeds are 100 rpm for Apparatus 1 (basket) and 50 rpm for Apparatus 2 (paddle) for solid-oral dosage forms and 25 rpm for suspensions. A 40-mesh screen is used in almost all baskets, but other mesh sizes may be used when the need is documented by supporting data.
Apparatus 2 is generally preferred for tablets. Apparatus 1 is generally preferred for capsules and for dosage forms that tend to float or that disintegrate slowly. A sinker, such as a few turns of platinum wire, may be used to prevent a capsule from floating. Other types of sinker devices that achieve minimal coverage of dosage form surface are commercially available. Where the use of a sinker device is employed, it is incumbent on the analyst to assure that the device used does not alter the dissolution characteristics of the dosage form.
Dissolution testing should be conducted on equipment that conforms to the requirements in the chapter Dissolution 711 and that has been calibrated with both the USP Salicylic Acid and Prednisone Calibrator Tablets. The method of analysis should be validated in accordance with the procedures given in the chapter Validation of Compendial Procedures 1225.
The test time is generally 30 to 60 minutes, with a single time point specification for pharmacopeial purposes. To allow for typical disintegration times, test times of less than 30 minutes should be based on demonstrated need. Industrial and regulatory concepts of product comparability and performance may require additional time points, and this may also be a feature required for product registration or approval. For registration purposes, a plot of percentage of drug dissolved versus time should be determined. Enough time points should be selected to characterize adequately the ascending and plateau phases of the dissolution curve.
Dissolution test times and specifications usually are established on the basis of an evaluation of dissolution profile data. Typical specifications for the amount of active ingredient dissolved, expressed as a percentage of the labeled content (Q), are in the range of 70% to 80% Q dissolved. A Q value in excess of 80% is not generally used, as allowance needs to be made for assay and content uniformity ranges.
For products containing more than a single active ingredient, dissolution normally should be determined for each active ingredient. Where a dissolution test is added to an existing monograph, the disintegration test is deleted. However, in the case of sublingual preparations, a short disintegration time may be retained as a monograph specification in addition to a dissolution requirement.
Dissolution and Drug Release Testing—Method Development for Modified-Release Dosage Forms
Drug release testing is required for all modified-release dosage forms in which absorption of the drug is necessary for the product to exert the desired therapeutic effect. The apparatus and procedure conform to the requirements and specifications given in the general chapter Drug Release 724.
The dissolution medium preferably is deaerated water or, if substantiated by the solubility characteristics of the drug or the formulation, buffered aqueous solutions (typically pH 4 to 8) or dilute acid (0.001 N to 0.1 N hydrochloric acid) may be used. (See above under Dissolution and Drug Release Testing—Method Development for Immediate-release Dosage Forms.) For modified-release dosage forms, the pH- and surfactant-dependence of the dosage form should be evaluated by in vitro testing in media of various compositions. The volume of medium will vary depending on the apparatus used and the formulation under test.
The choice of apparatus should be based on knowledge of the formulation design and actual dosage form performance in the in vitro test system. Apparatus 1 (basket) or Apparatus 2 (paddle) may be more useful at higher rotation frequencies (e.g., the paddle at 100 rpm). Apparatus 3 (reciprocating cylinder) has been found to be especially useful for bead-type modified-release dosage forms. Apparatus 4 (flow cell) may offer advantages for modified-release dosage forms that contain active ingredients having very limited solubility. Apparatus 7 (reciprocating disk) has been shown to have application to nondisintegrating oral modified-release dosage forms, as well as to transdermal dosage forms. Apparatus 5 (paddle over disk) and Apparatus 6 (cylinder) have been shown to be useful for evaluating and testing transdermal dosage forms.
At least three test times are chosen to characterize the in vitro drug release profile for Pharmacopeial purposes. Additional sampling times may be required for drug approval purposes. An early time point, usually 1 to 2 hours, is chosen to show that potential dose dumping is not probable. An intermediate time point is chosen to define the in vitro release profile of the dosage form, and a final time point is chosen to show essentially complete release of the drug. Test times and specifications are usually established on the basis of an evaluation of drug release profile data. For products containing more than a single active ingredient, drug release should be determined for each active ingredient.
Where a single set of specifications cannot be established to cover multisource monograph articles, application of a Case Three standard is appropriate. In Case Three, multiple drug release tests are included under the same monograph heading, and labeling requirements are included to indicate with which drug release test a specific product complies and, in some cases, the biological performance to be expected.
Drug release testing should be conducted on equipment that conforms to the requirements in the chapter Drug Release 724 and that has been calibrated with the appropriate USP calibrators. The method of analysis should be validated in accordance with the procedures given in the chapter Validation of Compendial Procedures 1225.
IN VIVO EVALUATION OF MODIFIED-RELEASE DOSAGE FORMS
In evaluating a modified-release product, a fundamental issue is the types of studies that should be performed to give reasonable assurance of safety and efficacy. While providing important information concerning the release characteristics of the drug from the dosage form, at present in vitro studies are most useful for such purposes as monitoring drug product stability and manufacturing process control. The assessment of safety and efficacy of a modified-release dosage form is best achieved through observing in vivo pharmacodynamics or pharmacokinetics. Moreover, where there is a well-defined, predictive relationship between the plasma concentrations of the drug or active metabolites and the clinical response (therapeutic and adverse), it may be possible to use plasma drug concentration data alone as a basis for the approval of a modified-release preparation that is designed to replace an immediate-release preparation.
The following guidelines are intended to provide guidance in drug substance evaluation and the design, conduct, and evaluation of studies involving modified-release dosage forms. While these guidelines will focus on oral drug delivery systems, the principles may be applicable to other routes of drug administration (e.g., transdermal, subcutaneous, intramuscular, etc.).
Characterization of Drug Substance
physicochemical properties
Physicochemical information necessary to characterize the drug substance in a modified-release dosage form should generally be no less than for the drug substance in an immediate-release dosage form. Additional physicochemical information may be needed on polymorphism, particle size distribution, solubility, dissolution rate, stability, and other release-controlling variables of the active drug entity under conditions that may react to the extremes of the physiologic environment experienced by the dosage form. For purposes of this chapter, active drug entity is taken to be the official drug substance.
pharmacokinetic properties
It is recommended to characterize thoroughly the input absorption profile of the active drug entity from a rapidly available preparation (an intravenous solution or oral solution or a well-characterized FDA-approved immediate-release drug product), which serves in turn as a reference to evaluate the input profile of the modified-release dosage form. This information together with the biological disposition characteristics for the active drug entity can characterize and predict changes in the bioavailability of the drug when input is modified as in the case of the modified-release dosage form. For example, if the active drug entity exhibits saturable first-pass hepatic metabolism, a reduction in systemic availability could result after oral administration if the input rate is decreased.
In designing an oral modified-release dosage form, it may be useful to determine the absorption of the active drug entity in various segments of the gastrointestinal tract (particularly in the colon in the case of dosage forms that may release drug in this region). The effects of food also may be important, and should be investigated.
disposition properties
The information required to characterize the processes of disposition of the active drug entity from a modified-release dosage form should include those generally determined for the same drug in an immediate-release dosage form. This may include the following:
- Disposition parameters—clearance, volume of distribution, half-life, mean residence time, or model-dependent or noncompartmental parameters.
- Linearity or characterization of nonlinearity over the dose or concentration range which could possibly be encountered.
- Accumulation.
- Metabolic profile and excretory organ dependence, with special attention to the active metabolites and active enantiomers of racemic mixtures.
- Enterohepatic circulation.
- Protein binding parameters and dialyzability.
- The effects of age, gender, race, and relevant disease states.
- Plasma/blood ratios.
- A narrow therapeutic index or a clinical response that varies significantly as a function of the time of day.
pharmacodynamic properties
Prior to developing a modified-release dosage form, information described below should be gathered.
Concentration-response relationships should be available over a dose range sufficiently wide to encompass important therapeutic and adverse responses. In addition, the equilibration time1 characteristics between plasma concentration and effect should be evaluated. These concentration-response relationships should be sufficiently characterized so that a reasonable prediction of the safety margin can be made if dose-dumping from the modified-release dosage form should occur. If there is a well-defined relationship between the plasma concentration of the active drug entity or active metabolites and the clinical response (therapeutic and adverse), the clinical performance of a new modified-release dosage form could be characterized by plasma concentration-time data. If such data are not available, clinical trials of the modified-release dosage form should be carried out with concurrent pharmacokinetic-pharmacodynamic measurements.
Characterization of the Dosage Form
physicochemical properties
The variables employed to characterize the physicochemical properties of the active drug entity as it exists or is discernible in the dosage form should be the same as those employed to characterize the drug substance. Solubility and dissolution profiles over a relevant pH range, usually from pH 1 to pH 7.4, should be obtained, with particular attention given to the effect of the formulation (as compared to the active drug entity). Characterization of formulations that are insoluble in aqueous systems may require the addition of sodium lauryl sulfate or other surfactant.
pharmacokinetic properties
The types of pharmacokinetic studies that should be conducted are a function of how much is known about the active drug entity, its clinical pharmacokinetic and biopharmaceutic properties, and whether pharmacokinetic studies are intended to be the sole basis for product approval. As a minimum, (1) a single-dose crossover study for each strength of a modified-release dosage form and (2) a multiple-dose, steady-state study using the highest strength of a modified-release dosage form are required to characterize the product. Some appropriate single-dose crossover and multiple-dose steady-state studies are described below.
In some modified-release capsule dosage forms, the strengths differ from each other only in the amount of identical beaded material contained in each capsule. In this case, a single-dose and a multiple-dose steady-state study at the highest dosage strength are sufficient. Other strengths may be characterized on the basis of comparative in vitro dissolution data.
The following pharmacokinetic studies would be needed for most modified-release dosage forms. Such studies may, in this instance, constitute the basis for characterization of the dosage form. If approval is to be sought without conducting clinical trials, it is recommended that there be preconsultation with the regulatory authorities to ensure that an adequate database exists for such approval.
The types of studies generally conducted can be categorized as follows.
Case A—
 study ii—
Where comparisons of the pharmacokinetic properties of an immediate-release dosage form at different doses are not available, or where the data show nonlinearity, steady-state crossover studies comparing effects of the modified-release dosage form with those of the immediate-release dosage form should be conducted at two different dose rates: one at the low end of the recommended dosing range and the second at the high end of the dosing range. In each case, the modified-release dosage form must meet the criteria described in Study I with respect to AUC and fluctuations in plasma drug concentrations. If there are significant differences between the modified-release dosage form and the immediate-release dosage form at either the low or the high dosing rate, these data alone are not adequate to characterize the product.
study ii—
Where comparisons of the pharmacokinetic properties of an immediate-release dosage form at different doses are not available, or where the data show nonlinearity, steady-state crossover studies comparing effects of the modified-release dosage form with those of the immediate-release dosage form should be conducted at two different dose rates: one at the low end of the recommended dosing range and the second at the high end of the dosing range. In each case, the modified-release dosage form must meet the criteria described in Study I with respect to AUC and fluctuations in plasma drug concentrations. If there are significant differences between the modified-release dosage form and the immediate-release dosage form at either the low or the high dosing rate, these data alone are not adequate to characterize the product.
Case A applies to the original modified-release oral dosage form of an active drug entity already marketed in immediate-release form and for which extensive pharmacodynamic/pharmacokinetic data exist.
Single-dose Crossover Study—
A single-dose crossover study should include the following treatments: the modified-release dosage form administered under fasting conditions; a dosage form that is rapidly available administered under fasting conditions; and the modified-release dosage form administered at the same time as a high-fat meal (or another type of meal that has potential for causing maximum perturbation).
The study of food effects should include provision for control of the fluid intake (e.g., 6 to 8 oz.) and temperature (e.g., ambient) at the time of drug administration. The dosage form should be administered within 5 minutes after completion of the meal.
If there are no significant differences in the rate or extent of bioavailability (AUC, Cmax, and Tmax) as a function of the meal, then additional food effect studies are not necessary.
If significant differences in bioavailability are found, it is necessary to define how food affects the modified-release dosage form, as well as how the food-drug effect relates to time.
The purpose of these studies is twofold: first, to determine whether there is a need for labeling instructions describing special conditions for administration with respect to meals and second, to provide information concerning the pattern of absorption of the modified-release dosage form compared to that of the immediate-release dosage form. The drug input function should be defined for modified-release dosage forms.2 This will aid in the development of an appropriate in vitro dissolution test. For dosage forms that exhibit high variability, replicate studies are recommended.
Use the following guidelines in evaluating food effect.
- If no well-controlled studies have previously defined the effects of a concurrent high-fat meal on an immediate-release dosage form, studies should be performed to determine whether a food effect is a result of problems with the dosage form, i.e., food-related changes in release, or food effects that are unrelated to the dosage form, such as changes in the drug's absorption from the gastrointestinal tract or changes in the drug's disposition (i.e., distribution or elimination) that are independent of absorption. The cause of the food effect should be determined by conducting a single-dose crossover study comparing the solution (or immediate-release dosage form) under fed and fasting conditions. If there is no effect of food, then one would conclude that there are problems with the dosage form. If there is an effect of food, then one would conclude that these are unrelated to the dosage form.
- The effect of timing on the food-drug effect should be tested by performing a four-way crossover study with the modified-release dosage form administered under the following treatment conditions: fasting, taken with a high-fat meal, 1 hour before a high-fat meal, and 2 hours after a high-fat meal.
- If the food effect on an immediate-release dosage form is determined to result from changes in the dissolved drug's absorption from the gastrointestinal tract or from changes in drug disposition, studies should be designed to define the appropriate relationship between drug dosing and meals.
- Alternative appropriate studies could be conducted if the applicant were to label the drug for administration with a meal that is not fat-loaded. In this case, an alternative meal composition should be considered.
- The entire single-dose, modified-release absorption profile should be monitored. Where appropriate (e.g., in a multiple-dose study) for specific drugs and drug delivery systems, blood samples should be taken following breakfast on the second day, before the second dose is administered. This sampling schedule is particularly important for once-a-day products.
- For delayed-release (enteric-coated) dosage forms, bioavailability studies to characterize adequately the food effects and to support the dosing claims stated in the labeling should be performed.
Multiple-dose, Steady-state Studies—
study i—
When data demonstrating linear pharmacokinetics exist for an immediate-release dosage form, a steady-state study should be conducted with the modified-release dosage form at one dose rate (preferably at the high end of the usual dose-rate range) using an immediate-release dosage form as a control. At least three trough-plasma drug concentration (Cmin) determinations should be made to ascertain that steady-state conditions have been achieved. Plasma-drug concentration determinations, over at least one dosing interval of the modified-release dosage form, should be made in each phase of the crossover study. It may be preferable (as in the case of rhythmic variation in absorption or disposition of the drug) to measure concentrations over an entire day in each phase. The presence or absence of circadian variation should be verified. The modified-release dosage form should produce an AUC that is equivalent to the immediate-release dosage form. The degree of fluctuation for the modified-release product should be the same as, or less than, that for the immediate-release dosage form given by the approved regimen. Appropriate concentration measurements should include unchanged drug and major active metabolites. For racemic drug entities, consideration should be given to the measurement of the active enantiomers [enantiomer/diasteriomer distinction].
Data can be misleading when obtained from subjects with atypical drug disposition or physiologic characteristics, relative to the target population. Therefore, subject selection should be randomized or from an appropriate target population. If the modified-release dosage form is for use in a specific subpopulation (e.g., for children), it should be tested in that population. Regardless of whether a drug exhibits linear or nonlinear pharmacokinetics, the basis for characterization is equivalence of AUC and of the relative degree of fluctuation of concentrations of the modified-release and immediate-release dosage forms.
Steady-state studies in selected patient population groups or drug interaction studies may also be necessary, depending upon the therapeutic use of the drug and the types of individuals for whom the modified-release dosage form will be recommended. For drugs having narrow therapeutic indices, it may be necessary to perform more extensive plasma concentration measurements to determine the potential for unusual drug-release patterns in certain subpopulations. In such studies, it is advisable to perform more than one AUC measurement per patient to assess variability with both the modified-release and the immediate-release dosage forms.
Case B—
Case B applies to a non-oral, modified-release dosage form of an already marketed active drug entity for which extensive pharmacodynamic/pharmacokinetic data exist.
Case A
studies (omitting the food effect studies) would be appropriate for the evaluation of a modified-release dosage form designed for a non-oral route of administration if the pattern of biotransformation to active metabolites is identical for the two routes. If the biotransformation patterns are different, then clinical efficacy studies should be performed with the modified-release dosage form. In addition, special studies may be necessary to assess specific risk factors related to the dosage form (e.g., irritation and/or sensitization at the site of application).
Case C—
Case C applies to a generic equivalent of an approved modified-release dosage form.
A generic equivalent of an approved modified-release dosage form should be bioequivalent to the standard modified-release dosage form in its rate and extent of availability (i.e., AUC, Cmax, Cmin, and degree of fluctuation) in crossover single-dose and steady-state studies. For an oral modified-release dosage form, the food studies described under Case A should also be performed.
Statistical Analysis
An appropriate statistical method should be selected. (See In Vivo Bioequivalence Guidances 1090).
The currently accepted criteria in the United States for equivalence for most dosage forms require that the mean pharmacokinetic parameters of the test dosage form should be within 80% to 125% of the reference dosage form using the 90% confidence interval (or, equivalently, the two-sided test procedure, P = 0.05), and the upper and lower bounds must be within the 90% confidence interval.
IN VITRO–IN VIVO CORRELATIONS
The term in vitro–in vivo correlation first appeared in pharmaceutical literature as a result of the awareness of the concepts of bioavailability and of in vitro dissolution rate determinations. The term in vitro–in vivo correlation refers to the establishment of a rational relationship between a biological property, or a parameter derived from a biological property produced by a dosage form, and a physicochemical property or characteristic of the same dosage form. The biological properties most commonly used are one or more pharmacokinetic parameters, such as Cmax or AUC, obtained following the administration of the dosage form. The physicochemical property most commonly used is a dosage form's in vitro dissolution behavior (e.g., percent of drug released under a given set of conditions). The relationship between the two properties, biological and physicochemical, is then expressed quantitatively.
With the proliferation of modified-release products, it becomes necessary to examine the concept of in vitro–in vivo correlation in greater depth. Unlike immediate-release dosage forms, modified-release products cannot be characterized using a single-time point dissolution test. Furthermore, with a modified-release product a patient is to experience a specific plasma level curve covering a finite time period, usually 12 to 24 hours. There must be some in vitro means of assuring that each batch of the same product will perform identically in vivo. An in vitro–in vivo correlation would satisfy this need. Initially it was thought that developing a meaningful correlation for immediate-release dosage forms would be an easier task than for modified-release products. However, because of the nature of the principles upon which each type is based, it is believed that an in vitro–in vivo correlation is more readily defined for modified-release dosage forms.
Modified-Release Dosage Forms
general considerations
Initial attempts at developing in vitro–in vivo correlations of modified-release products utilized the same concepts as those employed with immediate-release dosage forms. Thus, numerous attempts have been made to correlate one or more pharmacokinetic parameters, determined from in vivo studies of a product, with the amount released in a given time in an in vitro dissolution test. These were essentially single-point correlations. Such relationships might indicate that increasing or decreasing the in vitro dissolution rate of the modified-release dosage form would result in a corresponding directional change in the product's performance. However, they revealed little about the overall plasma level curve, which is a major factor for drug performance in the patient.
The recognition and utilization of deconvolution techniques as well as statistical moment calculations represented a major advance over the single-point approach in that these two methodologies utilize all of the dissolution and plasma level data available to develop the correlations. Therefore, there are at least three correlation techniques (i.e., deconvolution, statistical moment, and single point), available to the pharmaceutical scientist. There are marked differences in the quality of the correlation obtained with each procedure. Thus, these methods have been categorized and are discussed in terms of the advantages of each along with the resulting potential utility as a predictive tool for the pharmaceutical scientist.
CORRELATION LEVELS
Three correlation levels have been defined and categorized in descending order of usefulness. The concept of correlation level is based upon the ability of the correlation to reflect the entire plasma drug concentration–time curve that will result from administration of the given dosage form. It is the relationship of the entire in vitro dissolution curve to the entire plasma level curve that defines the correlation.
Level A—
This level is the highest category of correlation. It represents a point-to-point relationship between in vitro dissolution and the in vivo input rate of the drug from the dosage form. This latter factor is sometimes referred to as in vivo dissolution. In such a correlation, the in vitro dissolution and in vivo input rate curves are either directly superimposable or may be made to be superimposable by the use of a constant offset value. The mathematical description for both curves is the same. Such a procedure is most applicable to modified-release systems that demonstrate an in vitro release rate that is essentially independent of the typical dissolution media usually employed in pharmaceutics. However, this is not a requirement for a Level A correlation. With this correlative procedure, a product's in vitro dissolution curve is compared to its in vivo input curve (i.e., the curve produced by deconvolution of the plasma level data). This may be done by use of mass balance model-dependent techniques, such as the Wagner–Nelson procedure or the Loo–Riegelman method, or by model-independent, mathematical deconvolution.
The advantages of a Level A correlation are as follows:
- A point-to-point correlation is developed. This is not found with any other correlation level. It is developed using every plasma level and dissolution point that has been generated. Thus, it reflects the complete plasma level curve. As a result, in the case of a Level A correlation, an in vitro dissolution curve can serve as a surrogate for in vivo performance. Therefore, a change in manufacturing site, method of manufacture, raw material supplies, minor formulation modification, and even product strength using the same formulation can be justified without the need for additional human studies.
- A truly meaningful (in vivo indicating) quality control procedure, which is predictive of a dosage form's performance, is defined for the dosage form.
- The extremes of the in vitro quality control standards can be justified by a convolution or deconvolution procedure.
Level B—
Utilizes the principles of statistical moment analysis. The mean in vitro dissolution time is compared to either the mean residence time or the mean in vivo dissolution time. Like correlation Level A, Level B utilizes all of the in vitro and in vivo data but is not considered to be a point-to-point correlation because it does not reflect the actual in vivo plasma level curve, since there are a number of different in vivo curves that will produce similar mean residence time values. For this reason, unlike the case of a Level A correlation, one cannot rely upon a Level B correlation alone to justify formulation modification, manufacturing site change, excipient source change, etc. In addition, in vitro data from such a correlation could not be used to justify the extremes of quality control standards.
Level C—
This category relates one dissolution time point (t50%, t90%, etc.) to one pharmacokinetic parameter such as AUC, Cmax, or Tmax. It represents a single point correlation. It does not reflect the complete shape of the plasma level, which is the critical factor that defines the performance of modified-release products. Since this type of correlation is not predictive of actual in vivo product performance, it is generally only useful as a guide in formulation development or as a production quality control procedure. Because of its obvious limitations, a Level C correlation has limited usefulness in predicting in vivo drug performance and is subject to the same caveats as a Level B correlation in its ability to support product and site changes as well as justification of quality control standard extremes.
developing a correlation
This chapter does not define the only procedures for developing an in vitro–in vivo correlation, as any well-designed and scientifically valid approach would be acceptable. To assist the pharmaceutical scientist, one possible procedure for developing a Level A correlation is described below.
- The plasma level or urinary excretion data obtained in the definitive bioavailability study of the modified-release dosage form are treated by a deconvolution procedure. The resulting data may represent the drug input rate of the dosage form. It is also considered to represent in vivo dissolution when the rate-controlling step of the dosage form is its dissolution rate (i.e., drug absorption, after it has dissolved, is considered to be instantaneous). Any deconvolution procedure (i.e., mass balance or mathematical deconvolution) will produce acceptable results.
- The biobatch3 is subjected to in vitro dissolution evaluation, and the effect of varying the dissolution conditions investigated. Some of the variables that can be studied are the apparatus (it is preferable to use official apparatus), mixing intensity, and dissolution medium (pH, enzymes, surfactants, osmotic pressure, ionic strength, etc.). It is not always necessary to study the dosage form's dissolution behavior under all of the conditions indicated. The number of conditions investigated will depend largely on whether a correlation can be found with the in vitro results obtained under the more commonly investigated conditions such as apparatus, agitation intensity, or dissolution medium and pH value. Each formulation and every drug represents an individual challenge. The in vitro evaluation of the dosage form should be performed regardless of the correlation level being developed.
- The in vitro dissolution curve is then compared to the drug input rate curve. This can be performed by various methods. Simply positioning one curve on the other can often indicate the existence of a correlation. This may then be quantified by defining the equation for each curve and comparing the corresponding constants. The simplest way to demonstrate a correlation is to plot the fraction absorbed in vivo versus the fraction released in vitro. With a Level A correlation, this relationship is often linear with a slope of 1. The intercept may or may not be 0 depending upon whether there is a lag time before the system begins to release drug in vivo, or the absorption rate is not instantaneous resulting in the presence of some finite quantity of dissolved but unabsorbed drug. In either case, it is a point-to-point or a Level A correlation when the relationship is linear with a slope of 1. This indicates that the curves are essentially superimposable.
- If from the studies indicated in the in vitro dissolution evaluation above, the modified-release dosage form exhibits dissolution behavior that is independent of the variables studied, and a Level A correlation is demonstrated when the in vitro dissolution curve is compared to the drug input rate curve, it is likely that the correlation is general and can be extrapolated within a reasonable range for that formulation of the active drug entity. If, however, the dosage form exhibits dissolution behavior that varies with the in vitro conditions, it must be determined which set of dissolution conditions best correlates with in vivo performance. One can then establish whether the correlation is real or an artifact. This is achieved by preparing at least two formulations having significantly different in vitro behavior. One should demonstrate a more rapid release and the other a slower release than the biobatch. A pilot BA-BE study should be performed with these formulations, and the previously established correlation demonstrated for both. The formulation modifications of these batches should be based upon formulation factors that would be expected to influence the product's modified-release mechanism, and modification of these formulation factors are expected to influence the dosage form's release rate.
- Once a Level A correlation is established, it is possible that in vitro testing may be utilized for establishing the effects of manufacturing modifications such as minor formulation changes, manufacturing site and equipment change, alternative excipient suppliers, and a change in dosage form strength in the same formulation. It is questionable whether such an extrapolation with Level B and C correlations is possible.
Establishment of Dissolution Specification Ranges
It is relatively easy to establish a multipoint dissolution specification for a modified-release dosage form. The dissolution behavior of the biobatch may be used to define the amount to be released at each time point. The difficulty arises in the variation to be allowed around each time point. In the case of a Level A correlation, this may be done in two ways, both of which utilize the in vitro-in vivo correlation: convolution and deconvolution.
convolution
Reasonable upper and lower dissolution values are selected for each time point established from the biobatch. Historically, dissolution specifications have been selected by using the average dissolution of the development batches, with a range of ± 2.5 to 3 standard deviations. It is now expected that the average dissolution values are approximately the same as those of the biobatch. The dissolution curves defined by the upper and lower extremes are convoluted to project the anticipated plasma level curves that would result from administration of these formulations to the same panel to which the biobatch was administered. If the resulting plasma level data fall within the 95% confidence intervals obtained in the definitive BA-BE study, these ranges can be considered to be acceptable. An alternative acceptance approach that has been suggested is that when the therapeutic window for a drug has been defined, one may establish an upper and lower limit if the convolution results fall within the therapeutic window, even if they fall outside the confidence interval. If they fall outside the intervals, a more limited range must be established. This should be continued until the predicted values meet the desired ranges.
deconvolution
An acceptable set of plasma-level data is established both for a batch of material demonstrating a more rapid release and for one demonstrating a slower release than that of the biobatch. These may be selected by using the extremes of the 95% confidence intervals or ±1 standard deviation of the mean plasma level. These curves are then deconvoluted, and the resulting input rate curve is used to establish the upper and lower dissolution specifications at each time point.
In the case of Level B and C correlations, batches of product must be made at the proposed upper and lower limits of the dissolution range, and it must be demonstrated that these batches are acceptable by performing a BA-BE study.
Immediate-Release Dosage Forms
general considerations
Since the mechanisms for release of drug from modified-release dosage forms are more complex and variable than those associated with immediate-release dosage forms, it would be anticipated that an in vitro-in vivo correlation would be easier to develop with the later formulations. Unfortunately, most of the correlation efforts to date with immediate-release dosage forms have been based on the correlation Level C approach, although there also have been efforts employing statistical moment theory (Level B). Although it is conceivable that the same Level A correlation approach may be utilized with immediate-release dosage forms, until data have been gathered to support this concept, Level B and Level C are the best approaches that can be recommended with these dosage forms.
1
Equilibration time is a measure of the time-dependent discontinuity between measured plasma concentrations and measured effects. The discontinuity is more often characterized by the degree of hysteresis observed when the effect-concentration plot for increasing concentrations is compared with that for decreasing concentrations. Where the equilibration time is very short (i.e., rapid equilibration with no active metabolites generated), there will be little or no hysteresis. That is, the same effect will be observed for a given concentration independent of the interval between the time of dosing and the time that measurements are made.
2
Wagner–Nelson, Loo–Riegelman, and other deconvolution methods are found in textbooks on biopharmaceutics.
3
The batch that was used in the pivotal bioavailability study.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | William E. Brown
Senior Scientific Liaison 1-301-816-8380 |
(GCDF2010) General Chapters - Dosage Forms |
USP35–NF30 Page 663