Macromolecular substances can be obtained by a number of methods including extraction from natural sources, modification of naturally occurring protein, mammalian cell culture in vitro, mammalian cell culture in vivo, production by microorganisms, and chemical syntheses. From a compendial perspective, macromolecular articles derived from biotechnology processes—or more specifically from recombinant-DNA (rDNA) technology, hybridoma technology, and transformed continuous cell lines—are those articles for which official names have been established. These articles have official public standards for identity, strength (potency), quality, and purity. Advances in genetics and the applications of genetic engineering have made the production of new and existing macromolecular articles technologically and economically feasible.
The technologies involved in producing a protein by biotechnological processes have been widely documented and general guidelines have been established by the federal government. The products of biotechnology may be regulated as drugs, biologics, or diagnostics, depending on their source, composition, and intended use. The novel approaches permitted by biotechnology can make it difficult to apply classic definitions of these categories and FDA has advised manufacturers to seek clarification in the early stages of development for how a product will be regulated when classification is not obvious.1 The overall regulatory scheme for biotechnology-derived products is the same as for products in the same category produced by traditional manufacturing methods, with the addition of specific requirements suited to the biotechnology-derived product. The general requirements are described primarily in the applicable parts of the Code of Federal Regulations, Title 21. NIH has published a guideline for rDNA research that is mandatory for both public and private NIH-supported research. This guideline has wide acceptance and voluntary compliance is common by institutions and corporations not specifically governed by it.2 Laboratory safety practices, particularly protection from potentially infectious materials, are a concern.3 Producing macromolecular articles by biotechnological processes involves initially the cloning of a specific gene in the laboratory, or the construction of a synthetic gene, with subsequent insertion into a host cell and subcloning in a microorganism or cell culture; then a process development on a pilot scale to optimize yield and quality; and finally large-scale fermentation or cell culture processes. The next step, which is the most relevant to the development of compendial monographs, is the purification of the macromolecular proteins. This is followed by animal testing, clinical testing, regulatory approval, and marketing.
Development of relevant public standards for these macromolecular articles is generally closely linked to the processing technology used and the physicochemical and biological characteristics of a specific drug. Characterizations of these articles to ensure safety, purity, and activity should incorporate classical techniques as well as methods specific to the technology. There is always the possibility that these articles may cause some untoward effects in patients using them due to immunological sensitization as a result of a single (or multiple) molecular modification. Such a possibility requires precise characterization of these substances. Although it is theoretically possible to develop public standards for a macromolecular article, it is not possible to develop specific standards that incorporate all prospective methods of production. The compendial perspective is to develop public standards that can be applied to a final product without comprehensive knowledge of production details but which can ensure maintenance of safety, identity, strength, quality, and purity.
Testing for identity, purity, and activity generally requires the use of USP Reference Standards. It will be necessary to consider what USP Reference Standards might be required and how relevant they might be to the method of production as it relates to a final product's characteristics. Such decisions will be made on a product-by-product basis. Favorable consideration will be given to the use of USP Reference Standards that are representative of the specific products that have undergone clinical testing and are fully characterized.
Although early adoption in USP of general methods of analysis of macromolecular drugs could be conducive to early standardization of methods, the technology and analytical procedures are evolving very rapidly. Analytical procedures—chemical, physical, microbiological, and immunological—will be included in the specific product monographs.
SCOPE OF BIOTECHNOLOGY IN THE DEVELOPMENT OF PHARMACOPEIAL ARTICLES
Definition of Biotechnology—Historical Perspective
In its broadest definition, biotechnology refers to the use of living organisms, including isolated mammalian cells, in the production of products having beneficial use. This definition would place alcohol, antibiotic production, and dairy processing, for example, within the scope of biotechnology. However, the current interest in biotechnology is primarily a result of two major advances. The first advance was the development of rDNA technology, which allowed the genes of one species to be transplanted into another species. Thus, gene coding for the expression of a desired protein (usually human) could be inserted into a host prokaryotic or eukaryotic cell in such a manner that the host cell would then express usable quantities of the desired protein. The second major advance was the development of techniques for producing large quantities of monoclonal antibodies (i.e., antibodies arising from a single lymphocyte).
Biotechnology within the pharmaceutical industry generally refers either to the production of protein products using rDNA techniques or to the production of monoclonal antibodies. Other technologies, such as transgenic animals and plants, gene therapy, and antisense DNA, may have potential implications for the pharmaceutical industry in the future but are not within the scope of this chapter.
rDNA Technology
The major steps in the application of rDNA technology for production of a desired protein are outlined in this section. The critical first step is identification of the protein that is to be produced, followed by the isolation of the gene of interest (i.e., the DNA sequence coding for the desired protein). Once this gene is isolated and fully characterized, it is inserted into a suitable vector such as a plasmid, which is an extrachromosomal segment of DNA usually found in certain bacteria. The plasmid is then inserted into the host cell. Clones of the transformed host cell line are isolated, and those that produce the protein of interest in the desired quantities are preserved under suitable conditions as a cell bank. As manufacturing needs arise, the cloned cells can be scaled up in a fermentation or cell culture process to produce the protein product.
Although the rDNA process is more fully described elsewhere in this chapter, the following important points should be recognized. The vector (plasmid) generally contains a selectable marker that can be used to identify cells that contain this gene. This is in addition to the gene coding for the protein of interest and the regulatory nucleotide sequences necessary for plasmid replication and messenger RNA (mRNA) transcription (the first step in protein synthesis). Selection of the desired cells is simplified because only properly transformed cells containing the selectable marker gene will survive under the growth conditions used to identify and propagate the transformed cells. Typically, the bacterial and eukaryotic selectable markers may include both antibiotic resistance or genes that complement an auxotrophic host mutation. There are numerous examples of both types of markers in each system.
Significant differences exist in the rDNA production process between prokaryotic and eukaryotic cells. In general, bacterial cells express greater concentrations of protein product and require relatively simple media components. However, prokaryotic cells do not perform many important post-translational modifications such as glycosylation and, historically, it was not possible to express large proteins in E. coli. These limitations necessitate the use of eukaryotic cells in many cases. The production differences between eukaryotic and prokaryotic host cells have significant impacts that are reflected in the requirements for process validation, purification, and analytical methodology. These requirements are addressed later in this chapter.
Monoclonal Antibodies
Antibodies are proteins produced by differentiated B lymphocytes. Each lymphocyte produces an antibody of defined specificity (i.e., the antibody molecule recognizes a specific site or epitope on the antigen). Antibodies that are produced in immunized animals are formed from many different clones of B lymphocytes; hence, the name polyclonal antibodies. Because the harvest of blood from these animals, by definition, results in polyclonal antibody mixtures, the antisera have multiple epitope recognition sites with a wide variety of binding constants (avidity) and therefore vary from lot to lot. Antibodies that are produced by immortalized cell lines (hybridomas) derived from single B cells are referred to as monoclonal antibodies. The harvest of these cultures leads to an antibody of specific epitope recognition with a homogeneous binding constant.
B lymphocytes have a finite life span in culture and have to be immortalized to enable continuous monoclonal antibody production. At present, the most common procedure is through chemically-induced fusion of a mouse spleen cell with a mouse myeloma cell. The resultant mouse-mouse hybridoma cell inherits from the myeloma cell the ability to replicate continuously in culture and inherits from the spleen cell the ability to produce the desired monoclonal antibody. Cell banks of the hybridoma cell line can be used to produce a continuous supply of the monoclonal antibody, either in vivo (i.e., by injection into mice and subsequent collection of the ascites fluid), or in vitro (i.e., by conventional cell culture techniques). It should be mentioned that recent advances in molecular genetics have led to the development of transfectomas and E. coli- and bacteriophage-based production schemes that may offer advantages for future production of monoclonal antibodies.
Process validation, purification, and analytical considerations for monoclonal antibodies are conceptually similar to those for rDNA products. This is because both types of products are proteins and therefore require similar handling and assay procedures. Because monoclonal antibodies are the products of immortalized cell lines, there is concern that potential viral nucleic acid contaminants be effectively excluded or inactivated by the manufacturing processes, just as for recombinant products of continuous cell lines.
Commercial applications of monoclonal antibodies include both diagnostic and therapeutic uses. In some cases, the monoclonal antibody is coupled to another substance (e.g., an oncolytic agent, radionuclide, toxin), with the resultant antibody conjugate being the final product of interest. In this case, both the antibody intermediate and the final product require extensive process development and analytical characterization.
For the purposes of this chapter, the scope of biotechnology will be confined to rDNA and monoclonal antibody pharmaceutical products.
CHARACTERISTIC PRODUCTION PROCESSES
The major difference between biotechnology-derived products and other pharmaceutical products is the means of production used to generate the product. Biotechnology makes use of genetically modified living organisms to produce protein or peptidyl products. This statement is true for both rDNA-derived products as well as monoclonal antibody products. Biotechnology-derived products are therefore readily differentiated from proteins or peptides that have been obtained by isolation from natural source materials such as plasma, serum, or tissue, or by chemical synthesis.
Biotechnology-derived products are not significantly different from other protein pharmaceuticals after the protein purification process. Thus, the basic requirements for process validation, environmental control, aseptic manufacturing, and quality control/quality assurance systems are fundamentally the same for all pharmaceutical products. However, the complexity of these systems is often greater for biotechnology-derived products because the production of such bio-molecules generally requires highly developed cell propagation processes, complicated purification methods, and analytical control to ensure their homogeneity, lot-to-lot consistency, and safety.
This section describes in some detail only those significant factors that are unique to the processing of biotechnology-derived products. This includes descriptions of the various biological production systems now in use, and a discussion of purification issues.
rDNA Production
rDNA products are presently produced in prokaryotic (bacteria) or eukaryotic systems (e.g., yeast, mammalian cell culture). The choice of the production organism is generally a direct function of the molecular complexity of the protein that is to be produced as well as the economics and efficiency of the fermentation or cell culture process. The earliest biotechnology-derived products were produced in E. coli based on the high degree of understanding of its molecular biology. Within the last few years, however, the use of large-scale eukaryotic cell culture has become relatively commonplace.
prokaryotic (bacterial) production
Bacterial production of biotechnology-derived products offers a number of distinct advantages as well as certain disadvantages. As previously stated, the biology of bacteria is quite well understood and the safe and effective use of E. coli as the host organism for production has been well documented. Thus, the expression of a new protein in E. coli, if possible, is often easier to accomplish than in other, more theoretically suitable, expression systems. This may be offset, however, by the fact that E. coli produces proteins usually in a chemically reduced state. For proper folding, such proteins require the production of intramolecular disulfide bonds by oxidation. A second disadvantage is that all E. coli proteins begin their sequence with an N-formyl methionine residue that may not always be removed by E. coli proteolytic systems, thus possibly yielding a methionyl derivative of the desired natural protein. A third disadvantage of expression in E. coli is the potential for product degradation because of trace protease impurities. A fourth disadvantage is the requirement for endotoxin removal during purification. These limitations aside, the ease of use of E. coli and their generally high-expression yields for most proteins often have resulted in the continued preferential use of these bacteria, where feasible.
As previously described, the key element in rDNA technology is the recombinant plasmid, which contains the gene that codes for the protein of interest. Plasmids are simple and small circular extrachromosomal segments of bacterial DNA that are isolated from a bacterium and are self-replicating. The basic technology involves the specific enzymatic cleavage of a plasmid using endonucleases followed by the insertion of a new piece of DNA that contains the gene of interest. The resultant recombinant plasmid is considered the key raw material of rDNA technology. The recombinant plasmid is introduced into the host organism through a process called transformation, where it passes on its new genetic information and results in the production of the protein product. The large-scale growth of recombinant organisms can be conducted in commercial fermenters at scales in excess of 100,000 L, making these types of production systems extremely economical. There are, however, a number of issues that complicate E. coli fermentation systems. In some cases, the expressed protein product may cause cellular toxicity, and/or be extremely difficult to recover or purify because it may be sequestered into bacterial inclusion bodies as large semisoluble aggregates. Recent advances in E. coli molecular biology have led to the ability to express proteins into the periplasmic space, allowing for the removal of unwanted N-terminal methionine groups and leading to more readily purified proteins.
eukaryotic (mammalian cell and yeast) production
The development of eukaryotic cell culture for the production of vaccines has long been established in the pharmaceutical industry and an extensive database has been developed to ensure the suitability of such protein products in humans. The extension of this technology to rDNA products was primarily a response to the limitations in the use of E. coli. Particularly with respect to large proteins or glycoproteins, eukaryotic cell expression is an attractive alternative to a bacterial system because eukaryotic cells can secrete proteins that are properly folded and identical in primary, secondary, and tertiary structure to the natural human protein. Concerns about the economics of this production system orginally hindered its development. Recent advances, however, in improved expression levels, in large-scale cell culture using Chinese Hamster Ovary (CHO) cells, and in the formulation of more highly defined growth media have combined to dramatically improve the economic feasibility of eukaryotic cell substrates. The number of cell passages required for cloning, selection, amplification, and cell banking prior to production generally necessitates the use of immortal cell lines because nonimmortalized strains (i.e., diploid cultures) cannot be propagated long enough to provide an economically useful time in the production stage. Initial questions regarding the safety of such immortal cell lines were based on concerns over potential oncogenes and potential viral and retroviral contamination. These concerns have been minimized by the exhaustive analysis and characterization of master cell banks for adventitious (accidentally introduced) agents, by effective process validation studies, and by the safety data gathered to date for products produced by this method. The resultant thoroughly characterized master cell bank is used for full-scale production. Other eukaryotic cell lines, such as those derived from insect cells, may be useful in achieving many of the conformational and post-translational advantages that have been described for mammalian cell culture.
The use of yeast strains such as Saccharomyces cerevisiae for production has been extensively explored. The production of proteins in yeast offers many theoretical advantages over E. coli while raising certain new concerns. Like E. coli, yeast can maintain stable plasmids extrachromosomally; however, unlike E. coli, yeast possesses the ability to produce glycoproteins.
monoclonal antibody production
Monoclonal antibodies can be produced in two major ways, depending on whether they are of human or murine (mouse) origin. For antibodies of murine origin, appropriate lymphocytes are selected from the spleens of previously inoculated mice or rats. The cell is then fused with a transformed cell line such as a myeloma cell line, producing a hybridoma cell. The hybridoma cells are then clonally selected and used to produce the monoclonal antibody products. For antibodies of human origin, human B lymphocytes can be clonally selected for the hapten binding specificity of their product antibodies; these selected cells can then be immortalized by infection with a virus. The resultant fused or transformed cell can proliferate indefinitely in a bioreactor/cell culture environment or can be injected into mice from whose ascites fluid the protein can be obtained. Antibody is produced as directed by the chromosomal information that resides in the cell or was acquired during fusion and is secreted into the medium from which it can be readily purified. The hybridoma cells must be thoroughly analyzed and characterized in the same general way as an rDNA cell bank. The resultant cell bank is used for production of product either by large-scale cell culture or by harvesting ascites fluid from mice inoculated with transformed cells.
Control of Fermentation and Cell Culture Processes
Because the production process using a living system is the fundamental cornerstone of biotechnology, the issues that relate directly to the control of biotechnology processes need to be examined. Concerns over the production of proteins in bacteria, for example, primarily involve systems for ensuring genetic stability, consistent product yield, and evidence of the lack of contamination by adventitious organisms. These same concerns apply to large-scale eukaryotic cell culture, where, as stated above, there are also significant issues relating to the use of immortalized cell lines such as the putative presence of oncogenic DNA/RNA and impurities from media proteins.
fermentations (bacteria and yeast)
A considerable amount of knowledge has been obtained for the production of recombinant proteins in bacteria and yeast; therefore, the major fermentation issues typically are resolved by the demonstration of consistency in fermentation conditions. Fermentations with bacteria and yeast usually are performed over short, well-defined time periods to monitor and control growth rate and product expression conditions. The presence of contaminating foreign organisms may be detected by effects on growth rate, culture purity, fatty acid profile, etc., and is cause for termination of the fermentation. The genetic stability of the production plasmid for bacteria may be addressed by isolation and nucleotide sequence analysis or by DNA restriction mapping. These results may be confirmed by peptide mapping of the expressed protein for each product lot manufactured. It is very important to optimize the fermentation conditions so that the amount of proteolytic processing of the target protein that may occur can be either limited or avoided completely. Proteolytic processing is often a problem in E. coli fermentations and may lead to recovery difficulties and low product yields. Finally, the conformation of the protein and its effects on potency must be addressed by the fermentation process.
eukaryotic cell cultures
The origin of large-scale cell culture techniques for the production of biotechnology-derived products can be traced back to the vaccine industry. Developments such as large-scale cell suspension cultures using recombinant organisms that secrete the desired protein into the media have had a significant impact on biotechnology. Large glycosylated proteins in quantities sufficient for the marketplace can now be produced. The use of eukaryotic cell cultures, however, is complicated by issues such as genetic stability, protein folding, and culture conditions, including cell viability and growth rates. For example, the genetic stability of cell cultures cannot be addressed as readily as E. coli fermentations by techniques such as plasmid sequence analysis because the gene that codes for the product is incorporated into the cell genome and is not easily recovered. One alternative is peptide mapping of the expressed protein, which requires a resolution and sensitivity adequate to detect subtle mutations.
The absence of adventitious organisms in cell cultures is critical. In addition to demonstrating that bacteria, yeast, and molds are not present in cell cultures, the manufacturer must provide for each culture evidence that mycoplasmas and adventitious viruses are not present. It is important to recognize that certain hybridomas used for monoclonal antibody production may contain endogenous retroviruses. However, it must be demonstrated that any viruses present in the culture are removed from the final product. This requires the development of suitable analytical techniques to ensure the absence of contamination by mycoplasmas or human and animal adventitious viruses.
The degree and type of glycosylation may be important in the design of cell culture conditions for the production of glycosylated proteins. The degree of glycosylation present may affect the half-life of the product in vivo as well as its potency and antigenicity. Although the glycosylation status of a cell culture product is difficult to determine, it can be verified to be consistent if the culture conditions are highly reproducible.
Process for Recovery and Purification
The recovery of protein products obtained from either fermentation or cell culture is generally based on efficient protein separation techniques such as those listed in Table 1. The recovery process begins with isolation of the desired protein from the fermentation or cell culture medium, often in a very impure form. The advantage of cell culture and yeast-derived products is that many of these proteins are secreted directly into the medium, thus requiring only cell separation to obtain a significant purification. For E. coli-derived products, lysis of the bacteria is often necessary to recover the desired protein. It is important in each case to achieve rapid purification of the desired protein because proteases released by the lysed organisms may cleave the desired product. Such trace proteases are a major concern in the purification of biotechnology-derived products because they can be very difficult to remove, may complicate the recovery process, and can significantly affect final product stability.
Table 1. Chromatographic Purification Methods Used for
Biotechnology-derived Products
Biotechnology-derived Products
| Chromatofocusing |
| Reversed-phase chromatography |
| Hydrophobic interaction chromatography |
| Charge-transfer chromatography |
| Size-exclusion chromatography (molecular sizing) |
| Ion-exchange chromatography |
| Affinity chromatography |
The recovery process is usually designed to purify the final product to a high level. The purity requirement for a product depends on many factors, although chronic use products may be required to have much higher purity than those intended for single-use purposes. Biotechnology products contain certain impurities that the recovery processes are specifically designed to eliminate or minimize. These impurities include trace amounts of DNA, growth factors, residual host proteins, endotoxins, and residual cellular proteins from the media. The most common impurities of concern and suitable assay methods to detect them are presented in Table 2.
Table 2. Potential Impurities and Contaminants in Biotechnology-derived Products
| Impurities or Contaminants | Detection Method |
|---|---|
| Impurities | |
| Bacterial Endotoxins Test |
|
| SDS-PAGEa, Immunoassays | |
| SDS-PAGE, HPLCb, Immunoassays | |
| DNA hybridization, UV spectrophotometry, Protein binding | |
| Peptide mapping, HPLC, IEFc, MSd | |
| Peptide mapping, HPLC, MS | |
| Peptide mapping, amino acid analysis, HPLC, Edman degradation analysis, MS | |
| IEF, SDS-PAGE (reduced), HPLC, Edman degradation analysis | |
| SDS-PAGE, HPSECe | |
| IEF, HPLC, MS, Edman degradation analysis | |
| SDS-PAGE, immunoassays | |
| Amino acid analysis, peptide mapping, MS, Edman degradation analysis | |
| Contaminants | |
| Microbial Enumeration Tests |
|
| Modified 21 CFR Methodf, DNAFg | |
| CPEh and HAdi (exogenous virus only), reverse transcriptase activity, MAPj | |
|
a
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
b
High-performance liquid chromatography.
c
Isoelectric focusing.
d
Mass spectrometry.
e
High-performance size-exclusion chromatography.
f
Draft guidelines relating to Code of Federal Regulations, Title 21.
g
DNA-binding fluorochrome.
h
Cytopathic effect.
i
Hemadsorption.
j
Murine antibody production.
|
|
Chromatofocusing and reversed-phase chromatography are purification methods that use chemicals, either in the stationary (bonded) phase or in the mobile phase, that may become impurities in the final product. As in any new technology, the burden of validation (i.e., demonstrating removal of potentially harmful chemicals) is incumbent on the manufacturer. Validation is necessary when isolating end product monoclonal antibodies or using a technique that contains a monoclonal antibody purification step. The process must demonstrate removal of leaching antibody or antibody fragments. It is necessary to ensure the absence of adventitious agents such as viruses and mycoplasmas in the cell line that is the source of the monoclonal antibodies. The main concern is the possibility of contamination of the product with an antigenic substance whose administration could be detrimental to patients. Continuous monitoring of the process is necessary to avoid or limit such contamination. The problem of antigenicity related to the active as well as host proteins is one that is unique to biotechnology-derived products in contrast to traditional pharmaceuticals. Manufacturing methods that use certain solvents should be monitored if these solvents are able to cause chemical rearrangements that could alter the antigenic profile of the drug substance. The manufacturer is also obligated to produce evidence regarding performance consistency of novel chromatographic columns. Considerations for single-use products such as vaccines may differ because they are not administered continuously and, in this case, antigenicity is desirable. On the other hand, validating the removal of ligand or extraneous protein contamination is necessary. Unlike drugs derived from natural sources, manufacturers of biotechnology-derived products have been required to provide validation of the removal of nucleic acids during purification. Vaccines may again be different in this regard because of the accumulated clinical history on these products.
QUALITY CONTROL
In general, quality control systems for biotechnology-derived products are very similar to those quality control systems routinely employed for traditional pharmaceutical products in such areas as raw material testing and release, manufacturing and process control documentation, and aseptic processing. Quality control systems of biotechnology-derived products incorporate some of the same philosophies applied to the analysis of low molecular weight pharmaceutical products. These include the use of chemical reference standards and validated methods to evaluate a broad spectrum of known and/or potential product impurities and potential breakdown products. The quality control systems for biotechnology-derived products are generally analogous to those established for traditional biologicals with respect to determining product sterility, product safety in experimental animals, and product potency. For example, refer to Injections  1
1 , pH 791, Particulate Matter in Injections 788, Bacterial Endotoxins Test 85, and Impurities in Official Articles 1086.
, pH 791, Particulate Matter in Injections 788, Bacterial Endotoxins Test 85, and Impurities in Official Articles 1086.
The fundamental difference between quality control systems for biotechnology-derived products and traditional pharmaceuticals is in the types of methods that are used to determine product identity, consistency, purity, and impurity profiling. Furthermore, in biotechnology quality control, it is frequently necessary to use a combination of final product and validated in-process testing and process validation to ensure the removal of undesired real or potential impurities to the levels suggested by regulatory agencies. Biotechnology-derived products generally require a detailed characterization of the production organism (cell), a complete assessment of the means of cell growth/propagation, and explicit analysis of the final product recovery process.
The complexity of the quality control systems for biotechnology-derived products is related to both the size and structural characteristics of the product and manufacturing process. In general, the quality control systems required for products produced in prokaryotic cells are less complex than the systems required for products produced in eukaryotic cells. The quality control systems for prokaryotic production organisms usually entail documentation of the origin of the producer strain and encompass traditional testing for adventitious organisms, karyology, phenotyping, and antibiotic resistance. In addition, newer techniques such as DNA restriction mapping, DNA sequence analysis, and routine monitoring that may include measurement of mRNA and/or plasmid DNA levels may be useful. The quality control of the master cell bank and working cell bank for eukaryotic production organisms generally includes testing for adventitious organisms, karyology, identity, and stability monitoring. All eukaryotic cell lines (except yeast) are generally tested for the presence of retroviruses, retroviral activity markers, and tumorigenicity, although many of these tests may be of limited value.
PRODUCT FORMULATION
The products of biotechnology are proteins and peptides that are relatively unstable molecules compared to most organic pharmaceuticals. Most biotechnology processes involve the transfer of proteins from one stabilizing or solubilizing buffer to another during the purification process. Ultimately, the protein is exchanged into its final solution dosage form where long-term stability is achieved. In addition, these products often require lyophilization to achieve long-term stability because of the potential for degradation by a variety of mechanisms, including deamidation, aggregation, oxidation, and possible proteolysis by trace levels of host cell proteases. The final dosage form of the protein usually contains stabilizing compounds that result in the optimal pH and solution conditions necessary for long-term product stability and/or the desired properties for administration of the product (tonicity). These compounds include proteins, polyhydric alcohols, amino acids, carbohydrates, bulking agents, inorganic salts, and nonionic surfactants. In addition, these excipients may be required for stable lyophilized cake formation. There are special requirements for lyophilized products, such as the control of moisture levels, that generally are defined in the individual USP monograph and that may be important to product stability. Significantly, the assessment of protein stability usually requires the use of multiple analytical methods, each of which may be used to assess a specific mode of protein degradation. Many of these assays are described in the following section. The use of accelerated stability studies to predict the shelf life of protein formulations is often complicated by the effects of temperature on protein conformation, resulting in non-Arrhenius behavior. Thus, reliance on real-time, recommended storage condition stability studies is often required for establishing the expiration dating of biotechnology-derived products.
ANALYTICAL METHODOLOGY
The analysis of biotechnology-derived products relies heavily on the use of sophisticated analytical methods for demonstrating the structural identity and homogeneity of proteins and for evaluating the shelf life or stability of these products. This section discusses accuracy, precision, informational content, and general applicability of the most commonly used methods. Some methods, such as host cell impurity assays and residual DNA procedures, may be both highly process- and product-specific and thus should be included in the individual monographs.
Reference Standard Considerations
The use of suitable reference standards and/or reference materials is extremely important in the analysis of biotechnology-derived products. These standards may be either natural materials or proteins produced by genetic engineering. Many biotechnology-derived products require the availability of accurately characterized reference standards from internationally recognized sources such as the USP (see USP Reference Standards 11), WHO, NIH, and FDA. Currently, reference standards with defined activity units are available from these sources for some biologicals. These standards are used by the manufacturers in testing or to calibrate secondary standards using many of the assays described in this section. The potency value of the reference standard is obtained through collaborative studies that, when statistically evaluated, are used to determine the ultimate potency value assigned to the reference standard. The secondary standard can be used to determine the labeled amount of drug substance or potency defined on a product label. Thus, reference standards/reference materials for biotechnology-derived products that are used for the analytical purposes described in specific USP monographs will be approved and made available from USP. Ideally, these reference standards should be in use worldwide and should always be calibrated against the U.S. standard that is deposited by the manufacturer at FDA for those products licensed by FDA. This ensures the accurate and consistent determination of the activity, strength, and purity of these products. Because of a number of issues unique to biotechnology-derived products, such as process and product specificity, separate reference standards for similar products may be required. In addition, thorough development and recalibration of reference standards to replace depleted or expired standards will be conducted by USP to ensure that the label claims of the drug products do not change. One caveat in the assignment of the potency of the primary standard through collaborative studies is that units of activity so defined are only meaningful when compared in a single assay that is both suitably accurate and well described. Attempts to compare activity values from even subtly different assays can be expected to yield widely varying results.
Typical Methodology
There are a number of specific analytical methods that pertain to biotechnology-derived products. Many of the assays and tests described may be performed in different ways and, because some of these may be product specific as well, there is a need for clear guidelines on the application of specific methods to particular situations. See the chapters Design and Analysis of Biological Assays 111 and Validation of Compendial Procedures 1225 for some general information on methodology.
protein content
Protein content assays are used to quantitatively determine the amount of protein in a given biotechnology-derived product. The determination of protein content is often one of the most difficult measurements that needs to be made and often requires independent confirmation by alternate methods. Where applicable, methods such as UV spectrophotometry with a valid absorptivity and Kjeldahl nitrogen analysis can be used to determine absolute amounts of protein independent of reference standards. However, methods such as Lowry protein, biuret, and quantitative amino acid analysis, which require reference standards, also yield accurate values. Protein content assays are among the most important of all the methods used for these products because the results of other types of assays, such as potency, are also dependent on them.
There are several assays for the determination of protein content that are commonly used. These assays may be used at different points in the production process of a given biotechnology-derived product. For highly pure proteins, the simplest protein content method is based on the determination of the UV absorbance of a protein solution by spectrophotometry. The absorbance at the absorption maximum is determined and the protein concentration is calculated with the use of an empirically determined absorptivity. This technique is applicable to proteins containing the aromatic amino acid residues tryptophan, tyrosine, and/or phenylalanine. The absorption wavelength often used is 280 nm. The extinction coefficient, or molar absorptivity, should be determined in the same solvent that is used for the sample to be measured. If necessary, the product may be diluted prior to analysis to obtain solutions with absorbance values in the linear range of detection. Higher molecular weight aggregates and particulates may give rise to light-scattering effects, which provide artificially high absorbance values. Excipient components that have significant absorbance at 280 nm will also interfere with this test. UV spectrophotometry is unique among the protein content methods in that it is an absolute measure of concentration of a specific protein requiring no calibration with standards.
A commonly used general protein content method is the Lowry assay. This is based on the biuret reaction of proteins with copper (II) in a basic solution and the Folin-Ciocalteu phosphomolybdic-phosphotungstic acid reduction to heteropolymolybdenum blue by the copper-catalyzed oxidation of the aromatic amino acids tyrosine, tryptophan, and phenylalanine in the protein. The reaction products are blue and are quantitated spectrophotometrically in the visible region between 540 and 560 nm. This reaction is linear at microgram protein levels. The assay, however, is prone to interferences from a number of substances such as alcohols, sugars, and detergents. In some cases, interfering substances or product may be removed prior to analysis, e.g., by precipitation. Also, the preparation of controls containing interfering substances that are in the drug product may correct for their presence. Although bovine serum albumin historically has been used to prepare the standard curve, different proteins are known to react with differing intensity, so that a reference material of the same product should be used for calibration. The bicinchoninic acid (BCA) assay is a useful alternative to the Lowry assay because it is less sensitive to interfering substances. The working reagent is a BCA-copper (II) solution. The copper (II) complex is reduced to copper (I) in the presence of protein, and the purple color may be quantitated spectrophotometrically at approximately 560 nm.
Other colorimetric assays can also be used. The Bradford method, for example, employs the binding of the dye Coomassie Brilliant Blue to the protein in an acidic environment. The concentration of the protein in solution is then determined by comparing the absorbance at 595 nm with a standard curve of a reference material.
Fluorescent methods used are normally based on either fluorescamine or o-phthaldialdehyde (OPA). The main advantage of these assays is increased sensitivity. Another advantage is their use with hydrophobic proteins. Fluorescamine and OPA react with primary amines both at the N-terminus of the polypeptide and with amino acid side chains, such as lysine.
The Kjeldahl nitrogen method, Nitrogen Determination 461, provides an accurate and precise determination of protein concentration and is often used in the determination of UV protein absorptivities. The assay is performed in two stages. The sample is first decomposed with sulfuric acid to produce ammonium sulfate, carbon dioxide, and water. The decomposition is performed at the boiling point of sulfuric acid in long-necked, pear-shaped flasks. These flasks serve to condense water vapor and prevent the loss of material. Depending on the efficiency of decomposition, various salts such as potassium sulfate may be added to increase the boiling point of the sulfuric acid solution. Oxidizing agents such as perchloric acid or potassium permanganate have also been used to improve the decomposition. The second stage of the assay involves the direct determination of ammonia. In most macrodeterminations, ammonia is steam distilled from the mixture after basification with sodium hydroxide. The ammonia can typically be quantitatively distilled out of the mixture in 5 to 20 minutes and absorbed quantitatively into a standardized acidic solution of known volume and normality. The excess acid is then back-titrated with standardized base. For crude determinations of protein, the ammonia value (and therefore the nitrogen content), is multiplied by a factor of 6.25 mg of protein per mg of nitrogen, which corresponds to a nitrogen content of 16%. The protein value so obtained is generally valid for most proteins. If a more accurate value is required, as for an absorptivity determination, then the conversion factor must be calculated for the nitrogen content of the individual pure protein from the known amino acid composition. For glycoproteins that contain amino sugars, the calculated value is biased high unless a correction is applied.
Amino acid analysis is used in the determination of the appropriate absorptivity of the protein and may also be used quantitatively for the determination of protein content. This procedure, although more complicated than those described above, can also yield accurate results.
amino acid analysis
Amino acid analysis is a classical protein chemistry method for the determination of the amino acid composition of proteins and peptides. The method consists of the complete hydrolysis of a protein or peptide to its component amino acids, which are then chromatographically separated and quantitated. Amino acid analysis, therefore, can be used to determine both the amino acid composition of a product (i.e., identity) and the total amount of protein present. The method has some inherent difficulties, such as complete or partial destruction of some amino acids, that can be circumvented by appropriate analytical methodology. The amino acid tryptophan is destroyed by 6 N hydrochloric acid hydrolysis and thus requires the use of alternate hydrolysis conditions. The amino acids serine and threonine may be partially destroyed, whereas peptide bonds between bulky hydrophobic residues such as valine and isoleucine may be more resistant to hydrolysis, in both cases yielding values lower than actual. Accordingly, analysis of time-course hydrolysis samples may be used to compensate for these factors. Cysteine and methionine may require preoxidation to cysteic acid and methionine sulfone, respectively, for accurate quantitation. Each specific protein may require a procedure of optimized hydrolysis conditions for its amino acid analysis to obtain the optimal results.
Amino acid analysis is performed in two stages. The first stage involves the hydrolysis of the protein into its component amino acids. This hydrolysis is normally performed with 6 N hydrochloric acid at about 110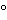 for 24 hours. Some proteins may require longer or more stringent hydrolysis conditions. The second stage is the separation and quantitation of the individual amino acids by some form of chromatography that can be performed with either precolumn or postcolumn derivatization. A number of precolumn derivative procedures are available, such as with OPA, phenylisothiocyanate (PITC), and fluorenylmethoxycarbonyl (FMOC). These derivatives are then separated by reversed-phase (RP) high-performance liquid chromatography (HPLC) and quantitated following UV or fluorescence detection. Postcolumn derivative methods involve separation of the component amino acids by high-performance ion-exchange chromatography (HPIEC) followed by postcolumn reaction with a chromophore, such as ninhydrin, and quantitation following UV/visible detection. All of these methods are suitable for performing amino acid analyses and each has its inherent advantages and disadvantages. OPA derivatives are very simple to prepare and are sensitive, requiring only a small amount of sample, but they are unstable and have to be chromatographed immediately upon preparation. Phenylthiocarbamyl (PTC) derivatives, on the other hand, are relatively more stable. Postcolumn derivatization with ninhydrin is often performed in the low-pressure mode and has the advantage of stability of the amino acid hydrolysate. Its disadvantage is the need for dual detection at 440 and 570 nm and for post-column apparatus.
for 24 hours. Some proteins may require longer or more stringent hydrolysis conditions. The second stage is the separation and quantitation of the individual amino acids by some form of chromatography that can be performed with either precolumn or postcolumn derivatization. A number of precolumn derivative procedures are available, such as with OPA, phenylisothiocyanate (PITC), and fluorenylmethoxycarbonyl (FMOC). These derivatives are then separated by reversed-phase (RP) high-performance liquid chromatography (HPLC) and quantitated following UV or fluorescence detection. Postcolumn derivative methods involve separation of the component amino acids by high-performance ion-exchange chromatography (HPIEC) followed by postcolumn reaction with a chromophore, such as ninhydrin, and quantitation following UV/visible detection. All of these methods are suitable for performing amino acid analyses and each has its inherent advantages and disadvantages. OPA derivatives are very simple to prepare and are sensitive, requiring only a small amount of sample, but they are unstable and have to be chromatographed immediately upon preparation. Phenylthiocarbamyl (PTC) derivatives, on the other hand, are relatively more stable. Postcolumn derivatization with ninhydrin is often performed in the low-pressure mode and has the advantage of stability of the amino acid hydrolysate. Its disadvantage is the need for dual detection at 440 and 570 nm and for post-column apparatus.
PROTEIN SEQUENCING
Protein sequencing is useful in the control of quality of protein biologicals because it can provide primary structure information, i.e., amino terminal and/or carboxy terminal structure. For rDNA-derived biologicals, this methodology has the additional purpose of confirming the complementary DNA (cDNA)-predicted amino acid sequence, protein homogeneity, and the potential extent of proteolytic clips. For monoclonal antibodies, this technique is used for determining protein homogeneity. Protein sequencing is divided into amino-terminal and carboxy-terminal sequencing applications and procedures.
Amino-Terminal Sequencing—
Amino-terminal sequence analysis is a classical protein chemistry technique that yields significant information about primary structure (sequence), homogeneity, and the presence of known or unknown cleavages in the polypeptide. N-terminal sequence analysis is performed with a number of commercially available automatic peptide sequencers. The method is based on the coupling reaction of the amino terminal residue of a protein or peptide with PITC. The resulting PTC-amino acid derivative is cleaved from the protein by a perfluoridated organic acid (generally trifluoroacetic or heptafluorobutyric acid), which exposes the adjacent amino acid. This next amino acid serves as a new N-terminus and is derivatized in the subsequent coupling and cleavage cycle. This process is repeated until an appropriate number, normally 8 to 10, of the amino acids are removed. The modified amino acid residue resulting from the cleavage cycle (anilinothiazolinone [ATZ]) is generally converted in the presence of acid and heat to a phenylthiohydantoin-amino acid (PTH-AA). The PTH-AA may then be determined following RP-HPLC analysis. Any intrachain cleavages as well as heterogeneity of the N-termini (e.g., N-terminal methionine) on the polypeptide will also be sequenced at the same time. These result in smaller peaks in the chromatogram and may enable both the relative quantitation of the amount of the N-termini and the identification of the location of the cleavage site on the polypeptide. This procedure for protein sequence analysis may also be performed manually. The limitations of the PITC sequencing method are that the method is only semiquantitative (i.e., the amount of the N-termini can only be estimated) and the PTH derivatives of serine and threonine may be severely degraded, making their determination difficult. Cysteine residues in order to be determined, must first be modified, for example by alkylation. In addition, the amino acids glycine and proline are slow to rearrange, resulting in minor difficulty in their determination.
Carboxy-Terminal Sequencing—
Sequencing of the protein from the carboxy terminus also yields valuable primary structure information as well as possible C-terminal cleavages. The sequential degradation of a protein from the C-terminus can be performed by either chemical or enzymatic methods. The reaction of hydrazine, ammonium thiocyanate, or cyanogen bromide with a protein can be used to sequentially degrade the protein at or near the C-terminus. The ammonium thiocyanate reaction has been extended for use on proteins coupled to solid supports. The C-terminal amino acids can be sequentially cleaved enzymatically with exopeptidases such as carboxypeptidases. Limitations of the carboxypeptidase approach are the potential contamination with endopeptidase and the inherent difficulty and unpredictable nature of the sequencing. Mass spectrometry can be used either directly on protein digests or in conjunction with HPLC peptide mapping to identify the C-terminus of the protein. However, these methods are only semiquantitative.
peptide mapping by hplc
For pharmaceutical proteins, peptide mapping has two primary purposes: it is a highly specific identity method and, in the case of biotechnology-derived products, may serve as a confirmation of genetic stability. Peptide mapping is used to compare the protein structure of a specific lot of material to that of a suitable reference material/reference standard or to those structures of previous lots to confirm correctness of the primary structure and to confirm lot-to-lot consistency of primary structure (within the limits of this technique). The amino- and carboxy-terminal peptides and carbohydrate-containing peptides often can be separated and identified. The latter are valuable in the peptide maps of glycosylated proteins such as monoclonal antibodies. Peptide mapping may be used to determine the presence of single or multiple incorrect amino acids that may result from such events as a single point mutation or mistranslation of the cDNA sequence.
The procedure involves the selective fragmentation of the protein into discrete peptides that are resolved by some chromatographic technique. The fragmentation is accomplished with endoproteases, such as trypsin, chymotrypsin, thermolysin, or V8 protease, or by selective chemical degradation with cyanogen bromide, which cleaves at specific sites on the molecule. Selection of the appropriate endoprotease to be used is directed by the primary sequence of the protein. Trypsin cleaves on the C-terminal side of the basic residues lysine and arginine; chymotrypsin cleaves after the aromatic residues phenylalanine, tyrosine, and tryptophan; thermolysin cleaves after the hydrophobic residues leucine, isoleucine, and valine; V8 protease cleaves after the acidic residues glutamic acid and aspartic acid; and cyanogen bromide cleaves at methionyl residues. Other enzymes, such as clostripain (arginine) and endoproteinase lys-C (lysine), and chemical methods, such as 2-nitro-5-thiocyanobenzoic acid (cysteine), may also be used. Each of these methods has its own set of advantages and disadvantages. One common disadvantage to all these techniques is that nonspecific cleavages occur to some degree. It is important that the peptides generated from the digestion are large enough to provide structural information about the protein, and yet small enough to allow their analysis and separation by a technique such as RP-HPLC. For this reason and the fact that cleavage with this enzyme is almost quantitative, trypsin is the enzyme with the most general applicability for most proteins. For large proteins of greater than 60,000 daltons (about 520 amino acids), cleavage with trypsin may result in too many fragments, so another endoprotease may be chosen. l-TPCK (tosyl-l-phenylalanine chloromethyl ketone)-treated trypsin normally is used because TPCK inhibits the action of chymotrypsin, a contaminant present in many trypsin preparations. Although reaction with cyanogen bromide cleaves at methionyl residues, proteins do not contain many of these residues. As a result, relatively few peptides are obtained and these may be too large or hydrophobic for HPLC separation.
Once the digestion is complete, the peptides are generally separated by either RP-HPLC and/or HPIEC. Selection of the appropriate column is empirically based and will vary for different proteins. For RP-HPLC, both 100-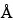 and 300- pore size supports work well, and the selection of the silica support may be an important criterion for optimal separation. For the smaller peptides generated by these digestions, C8 and C18 stationary phases generally have been found to be more efficient than C4 supports. The most common solvents used for reversed-phase separations are water and acetonitrile containing a constant (
and 300- pore size supports work well, and the selection of the silica support may be an important criterion for optimal separation. For the smaller peptides generated by these digestions, C8 and C18 stationary phases generally have been found to be more efficient than C4 supports. The most common solvents used for reversed-phase separations are water and acetonitrile containing a constant (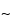 0.1%) amount of trifluoroacetic acid. Buffered mobile phases containing phosphate also offer excellent selectivity depending on the pH. Screening the effect of pH in the 3.0 to 5.0 range causes a shift of peptides containing the acidic residues, glutamic acid and aspartic acid. For ion-exchange separations, less information is available, but both silica and polymeric supports with both weak and strong ion-exchange stationary support can be used successfully. Because many of the peptides are somewhat hydrophobic, the addition of small amounts of organic solvents in the mobile phase, such as 5% to 10% methanol or acetonitrile, may be necessary. A potential disadvantage of HPIEC analysis of peptide mixtures is that sometimes neutral peptides or peptides that have the same charge as the support may not be retained on the column and thus may not be separated or identified by this method.
0.1%) amount of trifluoroacetic acid. Buffered mobile phases containing phosphate also offer excellent selectivity depending on the pH. Screening the effect of pH in the 3.0 to 5.0 range causes a shift of peptides containing the acidic residues, glutamic acid and aspartic acid. For ion-exchange separations, less information is available, but both silica and polymeric supports with both weak and strong ion-exchange stationary support can be used successfully. Because many of the peptides are somewhat hydrophobic, the addition of small amounts of organic solvents in the mobile phase, such as 5% to 10% methanol or acetonitrile, may be necessary. A potential disadvantage of HPIEC analysis of peptide mixtures is that sometimes neutral peptides or peptides that have the same charge as the support may not be retained on the column and thus may not be separated or identified by this method.
immunoassays
Immunoassays are used either as active drug substance methods to identify and quantitate the protein of interest or as impurity profile methods to detect and quantitate known host cell protein impurities. Because these protein impurities may represent a large number of potential impurities at trace levels rather than a single impurity, the immunoassays must be sensitive and selective to detect as many of these impurities as possible. Immunoassays that can measure these impurities to very low levels have been developed for E. coli proteins (ECPs) and CHO proteins. Immunoassays additionally may serve as potency assays for monoclonal antibodies using an appropriate antigen.
Immunoassays consist of a large group of assays that depend on specific high-affinity antibody:antigen interactions. These assays include the radioimmunoassays (RIAs) and the enzyme-linked immunosorbent assays (ELISAs). RIAs are performed in a liquid or solid phase using an unlabeled antibody directed against the radiolabeled protein of interest. The principle of the RIA is that the inhibition of binding of labeled antigen to unlabeled antibody by samples is compared to the inhibition by known standards, thus allowing quantitation of the protein of interest.
Immunoradiometric assays (IRMAs) or sandwich RIAs employ two antibody preparations that are used to sandwich the protein of interest. The first antibody is unlabeled and is directed against the protein, and the second antibody is radiolabeled and may be directed against the protein or the first antibody. The entire antibody:antigen complex is isolated and the amount of radioactivity, and, therefore, the protein of interest, is determined. The development of an RIA or IRMA for a biotechnology-derived product requires careful attention to production of the antisera, preparation of the labeled tracer, preparation of a suitable reference standard, and methods for the separation of free antigen from bound antigen.
The most commonly used ELISA format of trace impurity analysis is the sandwich ELISA that utilizes two antibody preparations like the IRMA, but without radiolabeling. The first antibody is unlabeled and the second antibody has an enzyme such as horseradish peroxidase (HRP) or alkaline phosphatase attached. Basically, the ELISA method consists of applying a layer of purified antibodies to the host cell proteins onto microtitration plates, followed by the protein product. The enzyme antibody conjugate is added and allowed to bind to the antibody-bound host cell proteins. An appropriate substrate is added for color development, which is analyzed with a spectrophotometric plate reader. Such a multiantigen ELISA requires a representative reference standard preparation of appropriate host cell protein impurities to serve as the immunogen for preparation of the antibodies used for the assay. This reference standard preparation is usually prepared from a manufacturing production run yielding all of the expected host cell proteins except the product protein. The total absence of the product protein is necessary in this preparation to avoid the production of antibodies to the product itself when the reference standard is used as an immunogen. Because of varying affinities of polyclonal antibodies to multiantigen preparations, the absolute accuracy of the multiantigen methods and the ability to detect every potential antigen cannot be guaranteed.
electrophoresis
Electrophoretic assays are among the most common and powerful of the assays used to evaluate protein purity and homogeneity. They are valuable not only for the initial evaluation and release of biotechnology-derived products but also as stability-indicating methods for detecting molecular or chemical changes in the molecule as a result of denaturation, aggregation, oxidation, deamidation, etc. The use of these methods is facilitated by their simplicity and their requirement of only microgram quantities of sample. The two types of electrophoretic assays most often used for biotechnology-derived products are sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and isoelectric focusing (IEF).
The SDS-PAGE method separates proteins primarily by their molecular weight because, in the presence of the anionic detergent SDS, a net negatively charged protein SDS complex is formed. The sample is first denatured in the detergent, which disrupts the noncovalent intramolecular and intermolecular bonds that hold proteins together, and then it is electrophoresed through a polyacrylamide gel support. Protein migration through the gel is proportional to size so that smaller proteins migrate faster through the gel than larger ones. Samples are often electrophoresed under both reduced and nonreduced conditions to determine if impurities of the same molecular weight or if intramolecular proteolytic cleavages of the protein of interest are present. Although nonreducing SDS-PAGE is commonly used to estimate the state of aggregation and/or oligomerization of the protein of interest, this method will only permit observation of aggregates or oligomers that are stable in the presence of SDS and the conditions used for sample preparation and electrophoresis. Proteins consisting of multiple chains held together by disulfide bonds are broken down and separated into their individual polypeptide chains. Sample detection following electrophoresis can be quantitative with densitometric analysis of Coomassie Brilliant Blue stain or qualitative, but with increased sensitivity in the nanogram range, with silver stain. Silver stain SDS-PAGE may also be performed quantitatively under suitable conditions. With proper validation, Coomassie Brilliant Blue staining and densitometry may also be used to give quantitative determination of polypeptides in the nanogram range. SDS-PAGE coupled with Coomassie Brilliant Blue stain is used to quantitatively determine the purity of the sample with regard to dimer and larger covalent aggregates and fragments. When the method is combined with the silver stain technique, an assessment of low/trace levels of a new impurity can be made by directly comparing the electrophoresed sample to the electrophoresed reference material or standard under reduced and nonreduced conditions. Generally, silver staining is used qualitatively because of potential major variations in binding of silver from protein to protein, and relatively inconsistent background on a routine basis. An estimation of the quantity of an impurity can be obtained by electrophoresis of a known amount of an internal standard such as bovine serum albumin or lesser dilutions of the protein of interest on other lanes of the same gel. The SDS-PAGE separation of a protein can be combined with an immunological method such as immunoblotting. The resulting Western blot is used to determine the identity of an electrophoretic band (i.e., product related or host cell protein impurity). After electrophoresis, the separated proteins are transferred onto a nitrocellulose or polyvinylidene difluoride (PVDF) membrane and reacted with the antibody of interest. Visualization of the complex is done with an enzymatically or radiolabeled antibody.
The IEF method separates proteins on the basis of their charge in an electrical field. The charges on a protein originate from various sources within its amino acid composition, such as protonated amino groups, unprotonated carboxyl groups, deprotonated sulfhydryl groups or tyrosine residues, oxidized cysteine residues, and deamidated residues. However, for each protein there is a pH at which the protein is isoelectric and these charges cancel each other, with the net charge being effectively zero. IEF is performed in the native state in a support of loose-pore polyacrylamide or agarose gel containing ampholytes (amphoteric low molecular weight ions) that set up a pH gradient because of their migration within the support matrix when an electrical field is applied. Simultaneously, in the presence of the electrical field, positively charged proteins migrate towards the cathode and negatively charged proteins migrate towards the anode. Migration stops when each protein reaches the pH value in the support gradient where its net charge is zero. This is the apparent pI or isoelectric point of the protein. Because the migration of a protein is dependent on its amino acid composition, altered forms of the protein and other proteins will migrate to different points on the support. IEF gels may be stained for protein visualization with either Coomassie Brilliant Blue or silver stains. IEF is employed as an identity tool or to ensure the homogeneity of a protein (e.g., monoclonal antibodies) as demonstrated by a banding pattern with the correct pI range. The method can also be used to evaluate the stability of a biological product. Protein deamidation (i.e., glutamine or asparagine residue deamidation) over time leading to the production of a new carboxylic acid group results in molecules with a more acidic pI. IEF can provide information on the state of glycosylation of glycoproteins such as monoclonal antibodies, which may appear as many bands because of changes in the apparent charge on the protein molecule as a result of the sialic acid residues. IEF gel patterns are usually more complicated to interpret than those of SDS-PAGE and interpretation may require many assumptions or subjective judgment.
High-performance capillary electrophoresis (HPCE), which offers the potential advantages of very high protein resolution, is being thoroughly investigated because of recent advances in the technology.
chromatographic methods
Chromatographic methods have long been used in the determination of the purity of small organic molecules and proteins such as insulin (see Chromatography 621) and in the determination of the active ingredient and/or excipient concentration of pharmaceutical products. Chromatographic methods are also very effective in the determination of the purity of recombinant pharmaceuticals. However, the chromatography of proteins is far more difficult because of multiple modes of interaction with the chromatographic support as a result of the size and/or shape, charge, and hydrophobicity of the proteins. The most common chromatographic methods used to profile recombinant proteins are RP-HPLC, HPIEC, size-exclusion chromatography (HPSEC), and hydrophobic interaction chromatography (HIC). These methods involve the separation of proteins and are used to determine the purity of drug substances as well as the levels of known impurities or degradation products. A complication with all column chromatographic methods is determining the mass balance between column load and column eluate. Nevertheless, HPLC techniques are valuable for determining the purity and strength of protein pharmaceuticals.
The most common RP-HPLC analyses are performed on columns containing a C4 or C8 stationary phase on a silica-based or polymeric support. C18 stationary phases are also used but more often with smaller peptides in an application such as peptide mapping. Supports with pore sizes of at least 300 are preferred for proteins of molecular weight greater than about 10,000. For most RP-HPLC analyses, the proteins are eluted with aqueous acetonitrile gradients and the trifluoroacetic acid is kept constant at 0.1%. Other buffers such as phosphate or tris(hydroxymethyl)aminomethane (Tris) are also used, where the pH may be adjusted for added selectivity to achieve optimal separations.
HPIEC is an important method for purity determination. These separations are based on changes in the charge of the molecule and are useful for identifying and quantitating in protein pharmaceuticals common impurities such as oxidized (primarily oxidized methionine) and deamidated forms (glutamine and asparagine) and clipped or truncated forms. Both strong and weak ion-exchange stationary phases on either silica or polymeric supports can be used. Cation-exchange chromatography may be performed on sulfopropyl-type resins and are effective in distinguishing oxidation and deamidation products. Proteins are typically loaded onto a column equilibrated with water or a weak buffer, and eluted with a salt gradient, such as 0 to 1 M sodium chloride.
HPSEC is a technique that may provide information on the levels of aggregation and fragmentation in a protein pharmaceutical. Depending on the information needed, the mobile phase may be native, containing an aqueous buffer such as 100 mM phosphate, pH 7, or it may be denaturing, containing a low level of a chaotrope or detergent such as 0.1% SDS. The analyses are performed isocratically, with detection typically between 210 and 220 nm depending on the buffer used. Detection at 280 nm may also be used but is less sensitive. Classical size-exclusion chromatography was performed on soft polymeric supports such as cross-linked dextrans, polyacrylamide, or agarose. These, however, are better suited for low-pressure applications. As a result, a number of supports with increased mechanical strength have been developed. Commercially available silica-based and cross-linked agarose supports are now commonly used. HPSEC is also useful for the determination of clipped forms of proteins. Clipped chains often remain attached through the disulfide bonds of cysteine residues. Treatment of the sample with a reducing agent such as dithiothreitol or mercaptoethanol will cleave the disulfide bond and separate the chains. The clipped chains may then be resolved from unclipped forms by HPSEC.
HIC provides separation of proteins based on differences in their hydrophobicity under mild adsorption and elution conditions that generally prevent denaturation and subsequent loss of biological activity. A stationary phase that is weakly hydrophobic is used with a buffered aqueous mobile phase and an initial high-salt concentration to adsorb the protein, which is then selectively eluted using a decreasing salt gradient. Interactions occur between nonpolar amino acid residues that are exposed on the surface of the protein and hydrophobic groups that are present on the chromatographic matrix. A number of silica-based and polymeric supports combined with weakly hydrophobic ligands, such as polyethers, phenyl ethers, or short alkyl chains, have been developed for use in HIC. This technique can be used in the analysis, purification, and characterization of more labile hydrophobic proteins. Protein retention and selectivity can be modified by control of variables such as salt type and concentration, pH and selective-ion effects, temperature and gradient design, as well as by careful selection of the stationary phase.
quantitative assays
Biomimetic assays (assays that mimic the biological effect of the product) are of major significance in the discussion of assays for biotechnology-derived products. These assays measure the activity of the product and ensure that it is efficacious. Essentially, there are three major types of quantitative assays: animal model assays, cell culture-based assays and in-vitro (physicochemical) assays. Each of these assays has application in the control of biological products. Regardless of the type of quantitative assay employed, it is desirable and, in some cases, necessary, to use a biomimetic assay.
Animal Model Assays—
Biomimetic assays in animal models have been developed for routine use. Although these assays have a relatively long history of use, they have several major disadvantages such as the large number of animals and appropriate animal facilities and handlers required, the high cost of analysis, the long analysis time (i.e., several days to weeks), and poor reproducibility of results. They are, however, in use mainly because a cell culture-based or in-vitro assay has not been developed and demonstrated to be of equal or greater value. An example of such an assay is that used for the determination of the activity of human growth hormone (somatrem and somatropin). The potency of human growth hormone is determined with a rat weight gain bioassay. Hypophysectomized female rats are monitored for weight gain over an 11-day period after daily injections with human growth hormone. The relative potency of the test sample is obtained by statistical comparison of the activity of the sample to that of a reference material/reference standard. Animal models can be used as bioidentity tests if and when appropriate in-vitro biological and/or physicochemical assays are developed for the measurement of potency of products.
Cell Culture-Based Bioassays—
This group of assays is comparatively easier to perform, gives results faster (1 to 3 days), and is considerably less expensive and less wasteful of resources than the animal model assays. Cell culture-based bioassays provide information on the effect of the biological product in a living system, but they are imprecise as a consequence of the variances of living cells but not as imprecise as an animal model assay. However, they can be automated and therefore can be repeated sufficiently to provide relatively reproducible and accurate results. An example of this type of assay is the measurement of antiviral activity of human 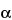 -interferon in a human diploid foreskin cell line or in a human lung carcinoma cell line (A549). This assay is performed in microtitration plates by incubation of cells with -interferon and subsequent challenge with encephalomyocarditis virus. The cells that survive are detected by dye binding and the dilution of -interferon where 50% protection of the monolayer occurs is calculated.
-interferon in a human diploid foreskin cell line or in a human lung carcinoma cell line (A549). This assay is performed in microtitration plates by incubation of cells with -interferon and subsequent challenge with encephalomyocarditis virus. The cells that survive are detected by dye binding and the dilution of -interferon where 50% protection of the monolayer occurs is calculated.
In Vitro (Physicochemical) Assays—
This group of assays does not rely on a living model, but is usually based on the chemical action of a biological product. These methods are comparatively simple, fast, precise, and accurate. The activity of tissue-type plasminogen activator (alteplase), for example, can be determined with an in vitro clot lysis assay that can be automated and can provide the required results within hours. A synthetic fibrin clot is formed in the presence of plasminogen as a result of the action of the enzyme thrombin on fibrinogen. When alteplase is added, the plasminogen is converted to the active enzyme plasmin, which then lyses the synthetic clot. The assay endpoint is followed spectrophotometrically or visually by noting the release of entrapped air bubbles. Another advantage of this type of assay, because of its precision and accuracy, is that it can be used to provide reliable estimates of the stability of the product. Examples of antibody:antigen and protein:ligand (receptor)-based in-vitro bioassays have also been developed for specific applications. These types of assays offer many advantages in their application to determine the potency of monoclonal antibodies or other highly ligand-specific proteins whose reactivity includes a binding step.
dna determination
Residual host cell DNA is a potential process-specific impurity in a biotechnology-derived product. The residual DNA is unique for each product because it is dependent on the host organism and the process recovery procedure used to manufacture the product. Although adverse health effects have not been reported from biologicals because of their DNA content, regulatory agencies have requested manufacturers to ensure that the DNA level in biotechnology-derived products is reduced to low levels.
The technique of DNA hybridization (dot blot analysis) is the most sensitive, routine DNA assay available to determine the DNA content of products. It is valuable as a purification process assay to demonstrate that a low level of DNA has been attained early in the manufacturing process. The method relies on the hybridization of cellular DNA from the sample with either specific 32P-labeled or chemically modified DNA probes obtained from the DNA of the host cell. The analysis is performed by first isolating any residual DNA in the sample by a procedure that may include hydrolysis of the protein, chromatography, organic extractions, and alcohol precipitation. The isolated DNA is denatured and then applied to a nitrocellulose or nylon membrane along with a set of serially-diluted host DNA standards. Positive and negative DNA controls are also applied and the membrane may be baked at approximately 80 or placed under UV light to complete binding of the DNA to the membrane. A DNA probe is then prepared either by nick translation, random primer synthesis, or chemical modification of a DNA extract of the host cell. The DNA probe is purified and thermally denatured at 95. It is then added to the baked or UV-treated membrane and allowed to hybridize with the DNA of the samples at approximately 42 in the presence of formamide or at higher temperatures without formamide for 24 to 48 hours. The membrane is subsequently placed between two X-ray films and exposed to produce an autoradiograph or is developed by immunochemical means using an enzyme conjugate/substrate system similar to ELISA and/or Western blot. The DNA of the sample is estimated by visual comparison of the dot intensity of the sample to those of the diluted DNA standards. The autoradiogram can also be scanned by optical densitometry. The sensitivity of the assay, i.e., 10 to 250 pg, is determined by the limit of visual detection above background of the serially-diluted DNA standards.
Other methods for DNA determination have been developed using biosensor technology. This methodology currently determines total DNA/nucleic acid impurities rather than specific host cell DNA. This technology may become quite valuable in the future, especially when more specific DNA binding methods are developed. Finally, the recently developed polymerase chain reaction (PCR) technology, which involves DNA amplification, may prove useful in detection and identification of contaminant DNA. Quantitative use of this technology, however, will require further development.
carbohydrate determination
One of the possible post-translational modifications that occurs on proteins is the covalent attachment of oligosaccharide chains. Glycosylation is a characteristic of recombinant proteins that are expressed from eukaryotic cell lines. Although the polypeptide chain of a glycoprotein is synthesized under the direct control of the genetic code, oligosaccharides are not primary gene products, but are synthesized by enzymes known as glycosyltransferases. This synthesis results in microheterogeneity of the carbohydrate chains. Also, glycosylation is cell-line dependent, so glycoproteins with identical polypeptide chains made in different cell lines may have considerably different carbohydrate structures. The sugars commonly found in glycoproteins include neutral sugars (d-galactose, d-mannose, and l-fucose), amino sugars (N-acetylglucosamine and N-acetylgalactosamine), and the acidic sugar, sialic acid.
Two main approaches can be taken to determine the sugars covalently attached to the glycoprotein. Both are based on the understanding that microheterogeneity is a common phenomenon among glycoproteins, and that the information represents either average composition or representative structures.
The first approach is the determination of the composition of sugars in a glycoprotein, which can be performed by several methods. Neutral sugars and sialic acid may be determined by simple colorimetric tests. Total neutral sugars can be determined following reaction with phenol and sulfuric acid and measuring the absorbance of the solution at about 490 nm compared to a standard curve. Following mild acid hydrolysis and periodate oxidation, free sialic acid content can be determined with thiobarbituric acid and the absorbance of the solution at about 550 nm compared to a standard curve. Individual neutral sugars can be determined following acid hydrolysis by several methods. Underivatized, they can be separated by HPIEC at high pH and quantitated by pulsed amperometric detection. They may also be converted to the alditol peracetates with acetic anhydride or to the aldononitrile acetates with hydroxylamine hydrochloride and pyridine prior to peracetylation, and the derivatives separated by gas chromatography.
The second approach in determining the carbohydrate composition is to release and separate individual oligosaccharide structures covalently attached to the glycoprotein. This requires an understanding of the types of structures attached. The attachment of sugars to proteins can occur in two major ways: through an O-glycosidic bond involving the hydroxyl group of serine, threonine, or modified amino acids such as hydroxylysine or hydroxyproline, or through the N-glycosidic bond of asparagine. O-linked oligosaccharides can be released from the protein following beta-elimination under alkaline conditions and reduction of the reducing end sugar with sodium borohydride. N-linked oligosaccharides can be released chemically by hydrazinolysis or enzymatically by one of a variety of specific glycosidases, such as endo H, endo F, or peptido-N-glycanase. The oligosaccharides can then be separated by HPIEC at high pH and quantitated with pulsed amperometric detection. This results in an oligosaccharide or carbohydrate map analogous to the peptide map for the protein.
adventitious and endogenous agent detection
Specific assays pertaining to biotechnology focus on the detection of bacteria, fungi, mycoplasma, and viruses. These reflect the possible contaminants that may occur in both bacterial fermentation and mammalian cell culture. Control is exerted in a variety of ways including characterization of the master seed bank and the working cell banks to ensure freedom from these contaminants, evaluation of raw materials, the design and operation of closed manufacturing systems, testing of production lots, and validation of specific manufacturing processes to ensure that contaminants would be inactivated or removed if present.
Freedom of final sterile dosage forms from bacteria and fungi is usually evaluated by tests for sterility as described in Sterility Tests 71. Mycoplasma assays are performed by standard cultivation methods employing aerobic and anaerobic incubation of solid medium in plates and semisolid broth in tubes and must comply with the code of federal regulations (21 CFR 610.12). In addition, noncultivable mycoplasma are detected microscopically by using the Hoechst bisbenzimide staining method.
Various methods that are used for the detection of adventitious virus contamination in cell lines include inoculation of indicator cell lines selected for their ability to support the replication of a broad range of viruses and monitoring these for markers of virus infection such as cytopathology, hemadsorption, hemagglutination, and immunofluorescence; inoculation of intact animals and monitoring for illness and death; inoculation of animals and, after four weeks, collection and evaluation of serum for antibodies to specific viruses of concern; and specific immunologic assays or genetic probes for some viruses of concern that cannot be detected by the other methods listed.
The expression of endogenous retrovirus genes is highly variable among different mammalian cells and cell lines. The unpredictable nature of their expression and the diversity of their biochemical and biological properties preclude the use of a single test and instead require an integrated testing strategy. Test methods generally used include transmission electron microscopy of cells from the master seed bank and ultracentrifuged pellets of cell-free, cell culture harvests; various assays for infectious retroviruses that use retrovirus-susceptible indicator cell lines; reverse transcriptase activity; and induction of retroviruses in cells of the master cell bank with chemicals known to induce retroviruses. In addition to classical virological methods, newer techniques such as molecular probe hybridization are also beginning to be used for these evaluations.
1
A series of documents entitled Points to Consider are available from the Director, FDA Center for Biologics Evaluation and Research, HFB-1, 8800 Rockville Pike, Bethesda, MD 20892.
2
This guideline was originally published in the Federal Register, Guidelines for Research Involving Recombinant DNA Molecules 1986; 51 (88): 16957-16985. Copies may be obtained from the Office of Recombinant DNA Activities, 12441 Parklawn Drive, Suite 58, Rockville, MD 20852.
3
A comprehensive guideline, Biosafety in Microbiological and Biomedical Laboratories, is available from the Superintendent of Documents, U.S. Government Printing Office, Washington, DC 20402, stock #107-040-000508-3.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Maura C Kibbey, Ph.D.
Senior Scientific Liaison, Biologics & Biotechnology 1-301-816-6309 |
(GCBA2010) General Chapters - Biological Analysis |
USP35–NF30 Page 491