Ritonavir
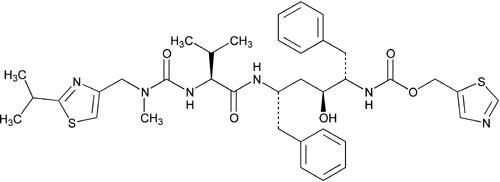
2,4,7,12-Tetraazatridecan-13-oic acid, 10-hydroxy-2-methyl-5-(1-methylethyl)-1-[2-(1-methylethyl)-4-thiazolyl]-3,6-dioxo-8,11-bis(phenylmethyl)-5-thiazolylmethyl ester [5S-(5R*,8R*,10R*,11R*)]-.
5-Thiazolylmethyl [(
» Ritonavir contains not less than 97.0 percent and not more than 102.0 percent of C37H48N6O5S2, calculated on the anhydrous basis.
Packaging and storage—
Preserve in tight, light-resistant containers. Store between 5 and 30.
and 30.
Identification—
A:
Infrared Absorption  197
197 —
—
Test specimen—
Dissolve 50 mg of Ritonavir in 1.0 mL of chloroform. Add 1 drop of this solution to the surface of a potassium bromide or a sodium chloride disk, and evaporate to dryness.
B:
The retention time of the major peak in the chromatogram of the Assay preparation is within 2% of the retention time of the major peak in the chromatogram of the Standard preparation, as obtained in the Assay.
X-ray diffraction 941
—The X-ray diffraction pattern conforms to that of USP Ritonavir RS if the drug substance is used for the solid dosage forms.
Heavy metals, Method II 231:
not more than 0.002%, using 1.0 g of Ritonavir and 2 mL of Standard Lead Solution (10 ppm Pb) in the Standard Preparation.
Water, Method I 921:
not more than 0.5%, determined on 0.500 g.
Residue on ignition 281:
not more than 0.2%, determined on 1.0 g.
Related compounds—
[note—Ritonavir is alkali sensitive. All glassware should be prerinsed with distilled water prior to use to remove residual detergent contamination.]
Monobasic potassium phosphate solution (0.03M), Diluent, Solution A, Solution B, and Mobile phase—
Prepare as directed in the Assay.
Standard stock solution and Intermediate standard solution—
Prepare as directed for Standard stock preparation and Intermediate standard preparation in the Assay.
Ritonavir identity standard solution—
Dissolve an accurately weighed quantity of USP Ritonavir Related Compounds Mixture RS, and dilute quantitatively with Diluent to obtain a solution having a concentration of about 1 mg per mL.
Standard solution—
Transfer 5.0 mL of the Intermediate standard solution to a 100-mL volumetric flask, dilute with Diluent to volume, and mix. [note—This solution may be used for 48 hours if stored at room temperature.]
Test solution—
Transfer about 50 mg of Ritonavir, accurately weighed, to a 50-mL volumetric flask. Dissolve in and dilute with Diluent to volume, and mix.
Chromatographic system (see Chromatography 621)—
The liquid chromatograph is equipped with a 240-nm detector and a 4.6-mm × 15-cm column that contains 3-µm packing L26 and is maintained at a constant temperature of about 60. The flow rate is about 1.0 mL per minute. The chromatograph is programmed as follows.
The run time for the Standard solution is 40 minutes, and the run time for the Test solution is 155 minutes. Chromatograph the Ritonavir identity standard solution and the Standard solution, and record the responses as directed for Procedure: the retention time of ritonavir is between 30 and 35 minutes; the resolution, R, between impurity E and impurity F (see Table 1) in the Ritonavir identity standard solution is not less than 1.0; the ratio of peak (Hp) to valley (Hv) of ritonavir and impurity N (regioisomer) is not less than 1; the capacity factor, k ¢, using the main component peak of the first Standard solution injection, is not less than 13; the column efficiency, using the main component peak of the first Standard solution injection, is not less than 5000 theoretical plates; the tailing factor, using the main component peak of the first Standard solution injection, is between 0.8 and 1.2; and the relative standard deviation of the peak area response of the main component peak, for replicate injections of the Standard solution, is not more than 3.0%.
| Time (minutes) |
Solution A
(%) |
Solution B
(%) |
Elution |
| 0 | 100 | 0 | equilibrium |
| 0–60 | 100 | 0 | isocratic |
| 60–120 | 100®0 | 0®100 | gradient |
| 120.1 | 0®100 | 100®0 | step gradient |
| 120.1–155 | 100 | 0 | isocratic |
Table 1. Approximate Relative Retention Time (RRT) for Known Related Impurities
| Impurity Identity | Common Name | Response Factor | RRT |
| A + B | Mixture of 2,4-Wing acid and monoacyl valine | — | 0.07 |
| C | Monoacylacetamide | — | 0.15 |
| D | 5-Wing diacyl | 1.37 | 0.24 |
| E | Oxidation impurity | — | 0.36 |
| F | Acid hydrolysis product | 0.73 | 0.39 |
| G | Ritonavir hydroperoxide | — | 0.45 |
| H | Acid/base by-product | 0.76 | 0.47 |
| I | Ethyl analog | — | 0.64 |
| J + K | Mixture of Boc-monoacyl and monoacyl isobutyl carbamate | 0.74 | 0.81 |
| L | Base cyclization product | 0.53 | 0.87 |
| M | 2,4-Wing isobutyl ester | — | 0.94 |
| N | Regioisomer | — | 1.05 |
| O | Isomer #2 | — | 1.11 |
| P | Di-monoacyl urea | — | 1.14 |
| Q | Isomer #4 | — | 1.23 |
| R | Isomer #1 | — | 1.32 |
| S | Di-monoacyl valine urea | — | 1.62 |
| T | 2,4-Wing diacyl | 0.73 | 2.87 |
| U | Triacyl impurity | — | 3.20 |
Procedure—
Separately inject equal volumes (about 50 µL) of the Diluent, Ritonavir identity standard solution, Standard solution, and Test solution into the chromatograph, record the chromatograms, and measure the peak area responses. Calculate the percentage of each impurity in the portion of Ritonavir taken by the formula:
100(CS / CT)(ri / rS)(1/F)P
in which CS and CT are the concentrations, in mg per mL, of ritonavir in the Standard solution and in the Test solution, respectively; ri is the peak response for each impurity obtained from the Test solution; rS is the average peak response of ritonavir obtained from the six injections of Standard solution; F is the response factor for each impurity, relative to ritonavir, as presented in Table 1; and P is the purity, in percentage, of USP Ritonavir RS taken to prepare the Standard solution: not more than 0.3% of impurity E and O is found; not more than 0.2% of impurity T is found; not more than 0.1% of any other impurity is found; and not more than 1.0% of total impurities is found.
Assay—
Monobasic potassium phosphate solution (0.03M)—
Dissolve about 8.2 of monobasic potassium phosphate in 2.0 L of water. Mix well, and filter through a 0.45-µm nylon membrane.
Diluent—
Prepare a mixture of Monobasic potassium phosphate solution (0.03M) and acetonitrile (1:1). Mix well, and filter through a 0.45-µm nylon membrane.
Solution A—
Prepare a mixture of the filtered Monobasic potassium phosphate solution (0.03M), acetonitrile, tetrahydrofuran (inhibitor-free), and n-butanol (69:18:8:5).
Solution B—
Prepare a mixture of acetonitrile, the filtered Monobasic potassium phosphate solution (0.03M), tetrahydrofuran (inhibitor-free), and n-butanol (47:40:8:5).
Mobile phase—
Use variable mixtures of Solution A and Solution B as directed for Chromatographic system. Make adjustments if necessary (see System Suitability under Chromatography 621). [note—Because of the high dependence of retention time and selectivity on the Mobile phase composition, the volumes should be accurately measured. Excessive or continued helium sparging must be avoided. Store the Mobile phase in a tightly sealed container when not in use.]
Standard stock preparation—
Transfer about 100 mg of USP Ritonavir RS, accurately weighed, to a 50-mL volumetric flask. Dissolve in and dilute with Diluent to volume, and mix. [note—This solution may be kept for 5 days if refrigerated.]
Intermediate standard preparation—
Transfer 5.0 mL of the Standard stock preparation to a 100-mL volumetric flask, dilute with Diluent to volume, and mix.
Standard preparation—
Transfer 25.0 mL of the Intermediate standard preparation to a 100-mL volumetric flask, dilute with Diluent to volume, and mix.
Assay preparation—
Transfer 5.0 mL of the Test solution, prepared as directed in the test for Related compounds, to a 50-mL volumetric flask, dilute with Diluent to volume, and mix. Dilute 25.0 mL of this solution with Diluent to 100-mL, and mix.
Chromatographic system—
Proceed as directed in the test for Related compounds. The run time for the Standard preparation and Assay preparation is 40 minutes. Chromatograph the Standard preparation, and record the responses as directed for Procedure: the capacity factor, k ¢, using the main component peak of the first Standard preparation injection, is not less than 13; the column efficiency, using the main component peak of the first Standard preparation injection, is not less than 5000 theoretical plates; the tailing factor, using the main component peak of the first Standard preparation injection, is between 0.8 and 1.2; and the relative standard deviation of the peak area response of the main component peak, for replicate injections of the Standard preparation, is not more than 2.0%.
Procedure—
Separately inject equal volumes (about 50 µL) of the Standard preparation and the Assay preparation into the chromatograph, record the chromatograms, and measure the peak area responses. Calculate the percentage, on the as-is basis, of C37H48N6O5S2 in the portion of Ritonavir taken by the formula:
0.5(WS / WT)( rT / rS)P
in which WS is the weight, in mg, of USP Ritonavir RS taken to prepare the Standard preparation; WT is the weight, in mg, of Ritonavir taken to prepare the Assay preparation; rT is the peak area of the impurity obtained from the chromatogram of the Assay preparation; rS is the average peak area of ritonavir obtained from the chromatograms of the five injections of the Standard preparation; and P is the purity, in percentage, of USP Ritonavir RS taken to prepare the Standard preparation.
Calculate the percentage, on the anhydrous basis, of C37H48N6O5S2 in the portion of Ritonavir taken by the formula:
100A/(100 – B)
in which A is the percentage of C37H48N6O5S2 on the as-is basis, as calculated above; and B is the percentage of water content.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
Chromatographic Column—
| Topic/Question | Contact | Expert Committee |
| Monograph | Behnam Davani, Ph.D., M.B.A.
Senior Scientist 1-301-816-8394 |
(MDAA05) Monograph Development-Antivirals and Antimicrobials |
| Reference Standards | Lili Wang, Technical Services Scientist 1-301-816-8129 RSTech@usp.org |
USP32–NF27 Page 3520
Pharmacopeial Forum: Volume No. 33(4) Page 679
Chromatographic columns text is not derived from, and not part of, USP 32 or NF 27.