INTRODUCTION
This chapter defines the terms and procedures used in chromatography and provides general information. Specific requirements for chromatographic procedures for drug substances and dosage forms, including adsorbent and developing solvents, are given in the individual monographs.
Chromatography is defined as a procedure by which solutes are separated by a dynamic differential migration process in a system consisting of two or more phases, one of which moves continuously in a given direction and in which the individual substances exhibit different mobilities by reason of differences in adsorption, partition, solubility, vapor pressure, molecular size, or ionic charge density. The individual substances thus separated can be identified or determined by analytical procedures.
The general chromatographic technique requires that a solute undergo distribution between two phases, one of them fixed (stationary phase), the other moving (mobile phase). It is the mobile phase that transfers the solute through the medium until it eventually emerges separated from other solutes that are eluted earlier or later. Generally, the solute is transported through the separation medium by means of a flowing stream of a liquid or a gaseous solvent known as the “eluant.” The stationary phase may act through adsorption, as in the case of adsorbents such as activated alumina and silica gel, or it may act by dissolving the solute, thus partitioning the latter between the stationary and mobile phases. In the latter process, a liquid coated onto an inert support, or chemically bonded onto silica gel, or directly onto the wall of a fused silica capillary, serves as the stationary phase. Partitioning is the predominant mechanism of separation in gas–liquid chromatography, paper chromatography, in forms of column chromatography, and in thin-layer chromatography designated as liquid-liquid chromatography. In practice, separations frequently result from a combination of adsorption and partitioning effects. Other separation principles include ion exchange, ion-pair formation, size exclusion, hydrophobic interaction, and chiral recognition.
The types of chromatography useful in qualitative and quantitative analysis that are employed in the USP procedures are column, gas, paper, thin-layer, (including high-performance thin-layer chromatography), and pressurized liquid chromatography (commonly called high-pressure or high-performance liquid chromatography). Paper and thin-layer chromatography are ordinarily more useful for purposes of identification, because of their convenience and simplicity. Column chromatography offers a wider choice of stationary phases and is useful for the separation of individual compounds, in quantity, from mixtures. Modern high-performance thin-layer chromatography, gas chromatography, and pressurized liquid chromatography require more elaborate apparatus but usually provide high resolution and identify and quantitate very small amounts of material.
Use of Reference Substances in Identity Tests—
In paper and thin-layer chromatography, the ratio of the distance (this distance being measured to the point of maximum intensity of the spot or zone) traveled on the medium by a given compound to the distance traveled by the front of the mobile phase, from the point of application of the test substance, is designated as the RF value of the compound. The ratio between the distances traveled by a given compound and a reference substance is the RR value. RF values vary with the experimental conditions, and thus identification is best accomplished where an authentic specimen of the compound in question is used as a reference substance on the same chromatogram.
For this purpose, chromatograms are prepared by applying on the thin-layer adsorbent or on the paper in a straight line, parallel to the edge of the chromatographic plate or paper, solutions of the substance to be identified, the authentic specimen, and a mixture of nearly equal amounts of the substance to be identified and the authentic specimen. Each sample application contains approximately the same quantity by weight of material to be chromatographed. If the substance to be identified and the authentic specimen are identical, all chromatograms agree in color and RF value and the mixed chromatogram yields a single spot; i.e., RR is 1.0.
Location and Identification of Components—
The spots produced by paper or thin-layer chromatography may be located by: (1) direct inspection if the compounds are visible under white or either short-wavelength (254 nm) or long-wavelength (360 nm) UV light, (2) inspection in white or UV light after treatment with reagents that will make the spots visible (reagents are most conveniently applied with an atomizer), (3) use of a Geiger-Müller counter or autoradiographic techniques in the case of the presence of radioactive substances, or (4) evidence resulting from stimulation or inhibition of bacterial growth by the placing of removed portions of the adsorbent and substance on inoculated media.
In open-column chromatography, in pressurized liquid chromatography performed under conditions of constant flow rate, and in gas chromatography, the retention time, t, defined as the time elapsed between sample injection and appearance of the peak concentration of the eluted sample zone, may be used as a parameter of identification. Solutions of the substance to be identified or derivatives thereof, of the reference compound, and of a mixture of equal amounts of these two are chromatographed successively on the same column under the same chromatographic conditions. Only one peak should be observed for the mixture. The ratio of the retention times of the test substance, the reference compound, and a mixture of these, to the retention time of an internal standard is called the relative retention time RR and is also used frequently as a parameter of identification.
The deviations of RR, RF, or t values measured for the test substance from the values obtained for the reference compound and mixture should not exceed the reliability estimates determined statistically from replicate assays of the reference compound.
Chromatographic identification by these methods under given conditions strongly indicates identity but does not constitute definitive identification. Coincidence of identity parameters under three to six different sets of chromatographic conditions (temperatures, column packings, adsorbents, eluants, developing solvents, various chemical derivatives, etc.) increases the probability that the test and reference substances are identical. However, many isomeric compounds cannot be separated. Specific and pertinent chemical, spectroscopic, or physicochemical identification of the eluted component combined with chromatographic identity is the most valid criterion of identification. For this purpose, the individual components separated by chromatography may be collected for further identification.
PAPER CHROMATOGRAPHY
In paper chromatography the adsorbent is a sheet of paper of suitable texture and thickness. Chromatographic separation may proceed through the action of a single liquid phase in a process analogous to adsorption chromatography in columns. Since the natural water content of the paper, or selective imbibition of a hydrophilic component of the liquid phase by the paper fibers, may be regarded as a stationary phase, a partitioning mechanism may contribute significantly to the separation.
Alternatively, a two-phase system may be used. The paper is impregnated with one of the phases, which then remains stationary (usually the more polar phase in the case of unmodified paper). The chromatogram is developed by slow passage of the other, mobile phase over the sheet. Development may be ascending, in which case the solvent is carried up the paper by capillary forces, or descending, in which case the solvent flow is also assisted by gravitational force.
Differences in the value of RF have been reported where chromatograms developed in the direction of the paper grain (machine direction) are compared with others developed at right angles to the grain; therefore, the orientation of paper grain with respect to solvent flow should be maintained constant in a series of chromatograms. (The machine direction is usually designated by the manufacturer on packages of chromatography paper.)
Descending Chromatography
In descending chromatography, the mobile phase flows downward on the chromatographic sheet.
Apparatus—
The essential equipment for descending chromatography consists of the following:
A vapor-tight chamber provided with inlets for addition of solvent or for releasing internal pressure. The chamber is constructed preferably of glass, stainless steel, or porcelain and is so designed as to permit observation of the progress of the chromatographic run without opening of the chamber. Tall glass cylinders are convenient if they are made vapor-tight with suitable covers and a sealing compound.
A rack of corrosion-resistant material about 5 cm shorter than the inside height of the chamber. The rack serves as a support for solvent troughs and for antisiphon rods which, in turn, hold up the chromatographic sheets.
One or more glass troughs capable of holding a volume of solvent greater than that needed for one chromatographic run. The troughs must also be longer than the width of the chromatographic sheets.
Heavy glass antisiphon rods to be supported by the rack and running outside of, parallel to, and slightly above the edge of the glass trough.
Chromatographic sheets of special filter paper at least 2.5 cm wide and not wider than the length of the troughs are cut to a length approximately equal to the height of the chamber. A fine pencil line is drawn horizontally across the filter paper at a distance from one end such that, when the sheet is suspended from the antisiphon rods with the upper end of the paper resting in the trough and the lower portion hanging free into the chamber, the line is located a few centimeters below the rods. Care is necessary to avoid contaminating the filter paper by excessive handling or by contact with dirty surfaces.
Procedure—
The substance or substances to be analyzed are dissolved in a suitable solvent. Convenient volumes, delivered from suitable micropipets, of the resulting solution, normally containing 1 to 20 µg of the compound, are placed in 6- to 10-mm spots not less than 3 cm apart along the pencil line. If the total volume to be applied would produce spots of a diameter greater than 6 to 10 mm, it is applied in separate portions to the same spot, each portion being allowed to dry before the next is added.
The spotted chromatographic sheet is suspended in the chamber by use of the antisiphon rod, which holds the upper end of the sheet in the solvent trough. The bottom of the chamber is covered with the prescribed solvent system. Saturation of the chamber with solvent vapor is facilitated by lining the inside walls with paper that is wetted with the prescribed solvent system. It is important to ensure that the portion of the sheet hanging below the rods is freely suspended in the chamber without touching the rack or the chamber walls or the fluid in the chamber. The chamber is sealed to allow equilibration (saturation) of the chamber and the paper with the solvent vapor. Any excess pressure is released as necessary. For large chambers, equilibration overnight may be necessary.
A volume of the mobile phase in excess of the volume required for complete development of the chromatogram is saturated with the immobile phase by shaking. After equilibration of the chamber, the prepared mobile solvent is introduced into the trough through the inlet. The inlet is closed and the mobile solvent phase is allowed to travel the desired distance down the paper. Precautions must be taken against allowing the solvent to run down the sheet when opening the chamber and removing the chromatogram. The location of the solvent front is quickly marked, and the sheets are dried.
The chromatogram is observed and measured directly or after suitable development to reveal the location of the spots of the isolated drug or drugs. The paper section(s) predetermined to contain the isolated drug(s) may be cut out and eluted by an appropriate solvent, and the solutions may be made up to a known volume and quantitatively analyzed by appropriate chemical or instrumental techniques. Similar procedures should be conducted with various amounts of similarly spotted reference standard on the same paper in the concentration range appropriate to prepare a valid calibration curve.
Ascending Chromatography
In ascending chromatography, the lower edge of the sheet (or strip) is dipped into the mobile phase to permit the mobile phase to rise on the chromatographic sheet by capillary action.
Apparatus—
The essential equipment for ascending chromatography is substantially the same as that described under Descending Chromatography.
Procedure—
The test materials are applied to the chromatographic sheets as directed under Descending Chromatography, and above the level to which the paper is dipped into the developing solvent. The bottom of the developing chamber is covered with the developing solvent system. If a two-phase system is used, both phases are added. It is also desirable to line the walls of the chamber with paper and to saturate this lining with the solvent system. Empty solvent troughs are placed on the bottom of the chamber, and the chromatographic sheets are suspended so that the end on which the spots have been added hangs free inside the empty trough.
The chamber is sealed, and equilibration is allowed to proceed as described under Descending Chromatography. Then the developing solvent (mobile phase) is added through the inlet to the trough in excess of the solvent required for complete moistening of the chromatographic sheet. The chamber is resealed. When the solvent front has reached the desired height, the chamber is opened and the sheet is removed and dried.
Quantitative analyses of the spots may be conducted as described under Descending Chromatography.
THIN-LAYER CHROMATOGRAPHY
In thin-layer chromatography, the adsorbent is a relatively thin, uniform layer of dry, finely powdered material applied to a glass, plastic, or metal sheet or plate, glass plates being most commonly employed. The coated plate can be considered an “open chromatographic column” and the separations achieved may be based upon adsorption, partition, or a combination of both effects, depending on the particular type of stationary phase, its preparation, and its use with different solvents. Thin-layer chromatography on ion-exchange layers can be used for the fractionation of polar compounds. Presumptive identification can be effected by observation of spots or zones of identical RF value and about equal magnitude obtained, respectively, with an unknown and a reference sample chromatographed on the same plate. A visual comparison of the size or intensity of the spots or zones may serve for semiquantitative estimation. Quantitative measurements are possible by means of densitometry (absorbance or fluorescence measurements), or the spots may be carefully removed from the plate, followed by elution with a suitable solvent and spectrophotometric measurement. For two-dimensional thin-layer chromatography, the chromatographed plate is turned at a right angle and again chromatographed, usually in another chamber equilibrated with a different solvent system.
Apparatus—
Acceptable apparatus and materials for thin-layer chromatography consist of the following.
A TLC or HPTLC plate. The chromatography is generally carried out using precoated plates or sheets (on glass, aluminum, or polyester support) of suitable size. It may be necessary to clean the plates prior to separation. This can be done by migration of, or immersion in, an appropriate solvent. The plates may also be impregnated by procedures such as development, immersion, or spraying. At the time of use, the plates may be activated, if necessary, by heating in an oven at 120 for 20 minutes. The stationary phase of TLC plates has an average particle size of 10–15 µm, and that of HPTLC plates an average particle size of 5 µm. Commercial plates with a preadsorbant zone can be used if they are specified in a monograph. Sample applied to the preabsorbant region develops into sharp, narrow bands at the preabsorbant-sorbent interface. Alternatively, flat glass plates of convenient size, typically 20 cm × 20 cm can be coated as described under Preparation of Chromatographic Plates.
for 20 minutes. The stationary phase of TLC plates has an average particle size of 10–15 µm, and that of HPTLC plates an average particle size of 5 µm. Commercial plates with a preadsorbant zone can be used if they are specified in a monograph. Sample applied to the preabsorbant region develops into sharp, narrow bands at the preabsorbant-sorbent interface. Alternatively, flat glass plates of convenient size, typically 20 cm × 20 cm can be coated as described under Preparation of Chromatographic Plates.
A suitable manual, semiautomatic, or automatic application device can be used to ensure proper positioning of the plate and proper transfer of the sample, with respect to volume and position, onto the plate. Alternatively, a template can be used to guide in manually placing the test spots at definite intervals, to mark distances as needed, and to aid in labeling the plates. For the proper application of the solutions, micropipets, microsyringes, or calibrated disposable capillaries are recommended.
For ascending development, a chromatographic chamber made of inert, transparent material and having the following specifications is used: a flat bottom or twin trough, a tightly fitted lid, and a size suitable for the plates. For horizontal development, the chamber is provided with a reservoir for the mobile phase, and it also contains a device for directing the mobile phase to the stationary phase.
Devices for transfer of reagents onto the plate by spraying, immersion, or exposure to vapor and devices to facilitate any necessary heating for visualization of the separated spots or zones.
A UV light source suitable for observations under short (254 nm) and long (365 nm) wavelength UV light.
A suitable device for documentation of the visualized chromatographic result.
Procedure—
Apply the prescribed volume of the test solution and the standard solution in sufficiently small portions to obtain circular spots of 2 to 5 mm in diameter (1 to 2 mm on HPTLC plates) or bands of 10 to 20 mm by 1 to 2 mm (5 to 10 mm by 0.5 to 1 mm on HPTLC plates) at an appropriate distance from the lower edge—during chromatography the application position must be at least 3 mm (HPTLC) or 5 mm (TLC) above the level of the developing solvent—and from the sides of the plate. Apply the solutions on a line parallel to the lower edge of the plate with an interval of at least 10 mm (5 mm on HPTLC plates) between the centers of spots or 4 mm (2 mm on HPTLC plates) between the edges of bands, and allow to dry.
Ascending Development—
Line at least one wall of the chromatographic chamber with filter paper. Pour into the chromatographic chamber a quantity of the mobile phase sufficient for the size of the chamber to give, after impregnation of the filter paper, a level of depth appropriate to the dimension of the plate used. For saturation of the chromatographic chamber, close the lid, and allow the system to equilibrate. Unless otherwise indicated, the chromatographic separation is performed in a saturated chamber.
Place the plate in the chamber, ensuring that the plate is as vertical as possible and that the spots or bands are above the surface of the mobile phase, and close the chamber. The stationary phase faces the inside of the chamber. Remove the plate when the mobile phase has moved over the prescribed distance. Dry the plate, and visualize the chromatograms as prescribed. For two-dimensional chromatography, dry the plates after the first development, and carry out a second development in a direction perpendicular to that of the first development.
Horizontal Development—
Introduce a sufficient quantity of the developing solvent into the reservoir of the chamber using a syringe or pipet. Place the plate horizontally in the chamber, connect the mobile phase direction device according to the manufacturer's instructions, and close the chamber. If prescribed, develop the plate starting simultaneously at both ends. Remove the plate when the mobile phase has moved over the distance prescribed in the monograph. Dry the plate, and visualize the chromatograms as prescribed.
For two-dimensional chromatography, dry the plates after the first development, and carry out a second development in a direction perpendicular to that of the first development.
Detection—
Observe the dry plate first under short-wavelength UV light (254 nm) and then under long-wavelength UV light (365 nm) or as stated in the monograph. If further directed, spray, immerse, or expose the plate to vapors of the specified reagent, heat the plate when required, observe, and compare the test chromatogram with the standard chromatogram. Document the plate after each observation. Measure and record the distance of each spot or zone from the point of origin, and indicate for each spot or zone the wavelength under which it was observed. Determine the RF values for the principal spots or zones (see Glossary of Symbols).
Quantitative Measurement—
Using appropriate instrumentation, substances separated by TLC and responding to ultraviolet-visible (UV-Vis) irradiation prior to or after derivatization can be determined directly on the plate. While moving the plate or the measuring device, the plate is examined by measuring the reflectance of the incident light. Similarly, fluorescence may be measured using an appropriate optical system. Substances containing radionuclides can be quantified in three ways: (1) directly by moving the plate alongside a suitable counter or vice versa; (2) by cutting the plates into strips and measuring the radioactivity on each individual strip using a suitable counter; or (3) by scraping off the stationary phase, dissolving it in a suitable scintillation cocktail, and measuring the radioactivity using a liquid scintillation counter (see Radioactivity  821
821 ).
).
The apparatus for direct quantitative measurement on the plate is a densitometer that is composed of a mechanical device to move the plate or the measuring device along the x-axis and the y-axis, a recorder, a suitable integrator or a computer; and, for substances responding to UV-Vis irradiation, a photometer with a source of light, an optical device capable of generating monochromatic light, and a photo cell of adequate sensitivity, all of which are used for the measurement of reflectance. In the case where fluorescence is measured, a suitable filter is also required to prevent the light used for excitation from reaching the photo cell while permitting the emitted light or specific portions thereof to pass. The linearity range of the counting device must be verified.
For quantitative tests, it is necessary to apply to the plate not fewer than three standard solutions of the substance to be examined, the concentrations of which span the expected value in the test solution (e.g., 80%, 100%, and 120%). Derivatize with the prescribed reagent, if necessary, and record the reflectance or fluorescence in the chromatograms obtained. Use the measured results for the calculation of the amount of substance in the test solution.
Preparation of Chromatographic Plates—
Apparatus—
Flat glass plates of convenient size, typically 20 cm × 20 cm.
An aligning tray or a flat surface upon which to align and rest the plates during the application of the adsorbent.
A storage rack to hold the prepared plates during drying and transportation. The rack holding the plates should be kept in a desiccator or be capable of being sealed in order to protect the plates from the environment after removal from the drying oven.
The adsorbent consists of finely divided adsorbent materials, normally 5 to 40 µm in diameter, suitable for chromatography. It can be applied directly to the glass plate or can be bonded to the plate by means of plaster of Paris [calcium sulfate hemihydrate (at a ratio of 5% to 15%)] or with starch paste or other binders. The plaster of Paris will not yield as hard a surface as will the starch, but it is not affected by strongly oxidizing spray reagents. The adsorbent may contain fluorescing material to aid in the visualization of spots that absorb UV light.
A spreader, which, when moved over the glass plate, will apply a uniform layer of adsorbent of desired thickness over the entire surface of the plate.
Procedure—
[note—In this procedure, use Purified Water that is obtained by distillation.] Clean the glass plates scrupulously, using an appropriate cleaning solution (see Cleaning Glass Apparatus 1051), rinsing them with copious quantities of water until the water runs off the plates without leaving any visible water or oily spots, then dry. It is important that the plates be completely free from lint and dust when the adsorbent is applied.
Arrange the plate or plates on the aligning tray, place a 5- × 20-cm plate adjacent to the front edge of the first square plate and another 5- × 20-cm plate adjacent to the rear edge of the last square, and secure all of the plates so that they will not slip during the application of the adsorbent. Position the spreader on the end plate opposite the raised end of the aligning tray. Mix 1 part of adsorbent with 2 parts of water (or in the ratio suggested by the supplier) by shaking vigorously for 30 seconds in a glass-stoppered conical flask, and transfer the slurry to the spreader. Usually 30 g of adsorbent and 60 mL of water are sufficient for five 20- × 20-cm plates. Complete the application of adsorbents using plaster of Paris binder within 2 minutes of the addition of the water, because thereafter the mixture begins to harden. Draw the spreader smoothly over the plates toward the raised end of the aligning tray, and remove the spreader when it is on the end plate next to the raised end of the aligning tray. (Wash away all traces of adsorbent from the spreader immediately after use.) Allow the plates to remain undisturbed for 5 minutes, then transfer the square plates, layer side up, to the storage rack, and dry at 105 for 30 minutes. Preferably place the rack at an angle in the drying oven to prevent the condensation of moisture on the back sides of plates in the rack. When the plates are dry, allow them to cool to room temperature, and inspect the uniformity of the distribution and the texture of the adsorbent layer; transmitted light will show uniformity of distribution, and reflected light will show uniformity of texture. Store the satisfactory plates over silica gel in a suitable chamber.
COLUMN CHROMATOGRAPHY
Apparatus—
The apparatus required for column chromatographic procedures is simple, consisting only of the chromatographic tube itself and a tamping rod, which may be needed to pack a pledget of glass wool or cotton, if needed, in the base of the tube and compress the adsorbent or slurry uniformly within the tube. In some cases a porous glass disk is sealed at the base of the tube in order to support the contents. The tube is cylindrical and is made of glass, unless another material is specified in the individual monograph. A smaller-diameter delivery tube is fused or otherwise attached by a leakproof joint to the lower end of the main tube. Column dimensions are variable; the dimensions of those commonly used in pharmaceutical analysis range from 10 to 30 mm in uniform inside diameter and 150 to 400 mm in length, exclusive of the delivery tube. The delivery tube, usually 3 to 6 mm in inside diameter, may include a stopcock for accurate control of the flow rate of solvents through the column. The tamping rod, a cylindrical ram firmly attached to a shaft, may be constructed of plastic, glass, stainless steel, or aluminum, unless another material is specified in the individual monograph. The shaft of the rod is substantially smaller in diameter than the column and is not less than 5 cm longer than the effective length of the column. The ram has a diameter about 1 mm smaller than the inside diameter of the column.
Column Adsorption Chromatography
The adsorbent (such as activated alumina or silica gel, calcined diatomaceous silica, or chromatographic purified siliceous earth) as a dry solid or as a slurry is packed into a glass or quartz chromatographic tube. A solution of the drug in a small amount of solvent is added to the top of the column and allowed to flow into the adsorbent. The drug principles are quantitatively removed from the solution and are adsorbed in a narrow transverse band at the top of the column. As additional solvent is allowed to flow through the column, either by gravity or by application of air pressure, each substance progresses down the column at a characteristic rate resulting in a spatial separation to give what is known as the chromatogram. The rate of movement for a given substance is affected by several variables, including the adsorptive power of the adsorbent and its particle size and surface area; the nature and polarity of the solvent; the hydrostatic head or applied pressure; and the temperature of the chromatographic system.
If the separated compounds are colored or if they fluoresce under UV light, the adsorbent column may be extruded and, by transverse cuts, the appropriate segments may then be isolated. The desired compounds are then extracted from each segment with a suitable solvent. If the compounds are colorless, they may be located by means of painting or spraying the extruded column with color-forming reagents. Chromatographed radioactive substances may be located by means of Geiger-Müller detectors or similar sensing and recording instruments. Clear plastic tubing made of a material such as nylon, which is inert to most solvents and transparent to short-wavelength UV light, may be packed with adsorbent and used as a chromatographic column. Such a column may be sliced with a sharp knife without removing the packing from the tubing. If a fluorescent adsorbent is used, the column may be marked under UV light in preparation for slicing.
A “flowing” chromatogram, which is extensively used, is obtained by a procedure in which solvents are allowed to flow through the column until the separated drug appears in the effluent solution, known as the “eluate.” The drug may be determined in the eluate by titration or by a spectrophotometric or colorimetric method, or the solvent may be evaporated, leaving the drug in more or less pure form. If a second drug principle is involved, it is eluted by continuing the first solvent or by passing a solvent of stronger eluting power through the column. The efficiency of the separation may be checked by obtaining a thin-layer chromatogram on the individual fractions.
A modified procedure for adding the mixture to the column is sometimes employed. The drug, in a solid form, and, as in the case of a powdered tablet, without separation from the excipients, is mixed with some of the adsorbent and added to the top of a column. The subsequent flow of solvent moves the drug down the column in the manner described.
Column Partition Chromatography
In partition chromatography the substances to be separated are partitioned between two immiscible liquids, one of which, the immobile phase, is adsorbed on a Solid Support, thereby presenting a very large surface area to the flowing solvent or mobile phase. The exceedingly high number of successive liquid-liquid contacts allows an efficiency of separation not achieved in ordinary liquid-liquid extraction.
The Solid Support is usually polar, and the adsorbed immobile phase more polar than the mobile phase. The Solid Support that is most widely used is chromatographic siliceous earth having a particle size suitable to permit proper flow of eluant.1 In reverse-phase partition chromatography the adsorbed immobile phase is less polar than the mobile phase and the solid adsorbent is rendered nonpolar by treatment with a silanizing agent, such as dichlorodimethylsilane, to give silanized chromatographic siliceous earth.
The sample to be chromatographed is usually introduced into the chromatographic system in one of two ways: (a) a solution of the sample in a small volume of the mobile phase is added to the top of the column; or, (b) a solution of the sample in a small volume of the immobile phase is mixed with the Solid Support and transferred to the column as a layer above a bed of a mixture of immobile phase with adsorbent.
Development and elution are accomplished with flowing solvent as before. The mobile solvent usually is saturated with the immobile solvent before use.
In conventional liquid-liquid partition chromatography, the degree of partition of a given compound between the two liquid phases is expressed by its partition or distribution coefficient. In the case of compounds that dissociate, distribution can be controlled by modifying the pH, dielectric constant, ionic strength, and other properties of the two phases. Selective elution of the components of a mixture can be achieved by successively changing the mobile phase to one that provides a more favorable partition coefficient, or by changing the pH of the immobile phase in situ with a mobile phase consisting of a solution of an appropriate acid or base in an organic solvent.
Unless otherwise specified in the individual monograph, assays and tests that employ column partition chromatography are performed according to the following general methods.
Solid Support—
Use purified siliceous earth. Use silanized chromatographic siliceous earth for reverse-phase partition chromatography.
Stationary Phase—
Use the solvent or solution specified in the individual monograph. If a mixture of liquids is to be used as the Stationary Phase, mix them prior to the introduction of the Solid Support.
Mobile Phase—
Use the solvent or solution specified in the individual monograph. Equilibrate it with water if the Stationary Phase is an aqueous solution; if the Stationary Phase is a polar organic fluid, equilibrate with that fluid.
Preparation of Chromatographic Column—
Unless otherwise specified in the individual monograph, the chromatographic tube is about 22 mm in inside diameter and 200 to 300 mm in length, without porous glass disk, to which is attached a delivery tube, without stopcock, about 4 mm in inside diameter and about 50 mm in length. Pack a pledget of fine glass wool in the base of the tube. Place the specified volume of Stationary Phase in a 100- to 250-mL beaker, add the specified amount of Solid Support, and mix to produce a homogeneous, fluffy mixture. Transfer this mixture to the chromatographic tube, and tamp, using gentle pressure, to obtain a uniform mass. If the specified amount of Solid Support is more than 3 g, transfer the mixture to the column in portions of approximately 2 g, and tamp each portion. If the assay or test requires a multisegment column, with a different Stationary Phase specified for each segment, tamp after the addition of each segment, and add each succeeding segment directly to the previous one.
If a solution of the analyte is incorporated in the Stationary Phase, complete the quantitative transfer to the chromatographic tube by scrubbing the beaker used for the preparation of the test mixture with a mixture of about 1 g of Solid Support and several drops of the solvent used to prepare the test solution.
Pack a pledget of fine glass wool above the completed column packing. The Mobile Phase flows through a properly packed column as a moderate stream or, if reverse-phase chromatography is applied, as a slow trickle.
Procedure—
Transfer the Mobile Phase to the column space above the column packing, and allow it to flow through the column under the influence of gravity. Rinse the tip of the chromatographic column with about 1 mL of Mobile Phase before each change in composition of Mobile Phase and after completion of the elution. If the analyte is introduced into the column as a solution in the Mobile Phase, allow it to pass completely into the column packing, then add Mobile Phase in several small portions, allowing each to drain completely, before adding the bulk of the Mobile Phase. Where the assay or test requires the use of multiple chromatographic columns mounted in series and the addition of Mobile Phase in divided portions is specified, allow each portion to drain completely through each column, and rinse the tip of each with Mobile Phase prior to the addition of each succeeding portion.
GAS CHROMATOGRAPHY
The distinguishing features of gas chromatography are a gaseous mobile phase and a solid or immobilized liquid stationary phase. Liquid stationary phases are available in packed or capillary columns. In the packed columns, the liquid phase is deposited on a finely divided, inert solid support, such as diatomaceous earth, porous polymer, or graphitized carbon, which is packed into a column that is typically 2 to 4 mm in internal diameter and 1 to 3 m in length. In capillary columns, which contain no packing, the liquid phase is deposited on the inner surface of the column and may be chemically bonded to it. In gas-solid chromatography, the solid phase is an active adsorbent, such as alumina, silica, or carbon, packed into a column. Polyaromatic porous resins, which are sometimes used in packed columns, are not coated with a liquid phase.
When a vaporized compound is introduced into the carrier gas and carried into the column, it is partitioned between the gas and stationary phases by a dynamic countercurrent distribution process. The compound is carried down the column by the carrier gas, retarded to a greater or lesser extent by sorption and desorption on the stationary phase. The elution of the compound is characterized by the partition ratio, k¢, a dimensionless quantity also called the capacity factor (see Glossary of Symbols for the definition of symbols). It is equivalent to the ratio of the time required for the compound to flow through the column (the retention time) to the elution time of an unretained compound. The value of the capacity factor depends on the chemical nature of the compound, the nature, amount, and surface area of the liquid phase, the column temperature, and the gas flow rate. Under a specified set of experimental conditions, a characteristic capacity factor exists for every compound. Separation by gas chromatography occurs only if the compounds concerned have different capacity factors.
Apparatus—
A gas chromatograph consists of a carrier gas source, an injection port, column, detector, and recording device. The injection port, column, and detector are temperature-controlled. The typical carrier gas is helium, nitrogen, or hydrogen, depending on the column and detector in use. The gas is supplied from a high-pressure cylinder or high-purity gas generator and passes through suitable pressure-reducing valves and a flow meter to the injection port and column. Compounds to be chromatographed, either in solution or as gases, are injected into the gas stream at the injection port. Depending upon the configuration of the apparatus, the test mixture may be injected directly into the column or be vaporized in the injection port and mixed into the flowing carrier gas prior to entering the column.
Once in the column, compounds in the test mixture are separated by virtue of differences in their capacity factors, which in turn depend upon vapor pressure and degree of interaction with the stationary phase. The capacity factor, which governs resolution, retention times, and column efficiencies of components of the test mixture, is also temperature-dependent. The use of temperature-programmable column ovens takes advantage of this dependence to achieve efficient separation of compounds differing widely in vapor pressure.
As resolved compounds emerge separately from the column, they pass through a differential detector, which responds to the amount of each compound present. The type of detector to be used depends upon the nature of the compounds to be analyzed and is specified in the individual monograph. Detectors are heated to prevent condensation of the eluting compounds.
Detector output is recorded as a function of time, producing a chromatogram, which consists of a series of peaks on a time axis. Each peak represents a compound in the vaporized test mixture, although some peaks may overlap. The elution time is a characteristic of an individual compound; and the instrument response, measured as peak area or peak height, is a function of the amount present.
Injectors—Sample injection devices range from simple syringes to fully programmable automatic injectors. The amount of sample that can be injected into a capillary column without overloading is small compared to the amount that can be injected into packed columns, and may be less than the smallest amount that can be manipulated satisfactorily by syringe. Capillary columns, therefore, often are used with injectors able to split samples into two fractions, a small one that enters the column and a large one that goes to waste. Such injectors may be used in a splitless mode for analyses of trace or minor components.
Purge and trap injectors are equipped with a sparging device by which volatile compounds in solution are carried into a low-temperature trap. When sparging is complete, trapped compounds are desorbed into the carrier gas by rapid heating of the temperature-programmable trap.
Headspace injectors are equipped with a thermostatically controlled sample heating chamber. Solid or liquid samples in tightly closed containers are heated in the chamber for a fixed period of time, allowing the volatile components in the sample to reach an equilibrium between the nongaseous phase and the gaseous or headspace phase.
After this equilibrium has been established, the injector automatically introduces a fixed amount of the headspace in the sample container into the gas chromatograph.
Columns—Capillary columns, which are usually made of fused silica, are typically 0.2 to 0.53 mm in internal diameter and 5 to 60 m in length. The liquid or stationary phase, which is sometimes chemically bonded to the inner surface, is 0.1 to 1.0 µm thick, although nonpolar stationary phases may be up to 5 µm thick. A list of liquid phases in current use is given in the section Chromatographic Reagents.
Packed columns, made of glass or metal, are 1 to 3 m in length with internal diameters of 2 to 4 mm. Those used for analysis typically are porous polymers or solid supports with liquid phase loadings of about 5% (w/w). High-capacity columns, with liquid phase loadings of about 20% (w/w), are used for large test specimens and for the determination of low molecular weight compounds such as water. The capacity required influences the choice of solid support.
Supports for analysis of polar compounds on low-capacity, low-polarity liquid phase columns must be inert to avoid peak tailing. The reactivity of support materials can be reduced by silanizing prior to coating with liquid phase. Acid-washed, flux-calcined diatomaceous earth is often used for drug analysis. Support materials are available in various mesh sizes, with 80- to 100-mesh and 100- to 120-mesh being most commonly used with 2- to 4-mm columns. Supports and liquid phases are listed in the section Chromatographic Reagents.
Retention time and the peak efficiency depend on the carrier gas flow rate; retention time is also directly proportional to column length, while resolution is proportional to the square root of the column length. For packed columns, the carrier gas flow rate is usually expressed in mL per minute at atmospheric pressure and room temperature. It is measured at the detector outlet with a flowmeter while the column is at operating temperature. The linear flow rate through a packed column is inversely proportional to the square of the column diameter for a given flow volume. Flow rates of 60 mL per minute in a 4-mm column and 15 mL per minute in a 2-mm column give identical linear flow rates and thus similar retention times. Unless otherwise specified in the individual monograph, flow rates for packed columns are about 30 to 60 mL per minute. For capillary columns, linear flow velocity is often used instead of flow rate. This is conveniently determined from the length of the column and the retention time of a dilute methane sample, provided a flame-ionization detector is in use. At high operating temperatures there is sufficient vapor pressure to result in a gradual loss of liquid phase, a process called bleeding.
Detectors—Flame-ionization detectors are used for most pharmaceutical analyses, with lesser use made of thermal conductivity, electron-capture, nitrogen-phosphorus, and mass spectrometric detectors. For quantitative analyses, detectors must have a wide linear dynamic range: the response must be directly proportional to the amount of compound present in the detector over a wide range of concentrations. Flame-ionization detectors have a wide linear range and are sensitive to most organic compounds. Detector response depends on the structure and concentration of the compound and on the flow rates of the combustion, air, makeup, and carrier gases. Unless otherwise specified in individual monographs, flame-ionization detectors with either helium or nitrogen carrier gas are to be used for packed columns, and helium or hydrogen is used for capillary columns.
The thermal conductivity detector employs a heated wire placed in the carrier gas stream. When an analyte enters the detector with the carrier gas, the difference in thermal conductivity of the gas stream (carrier and sample components) relative to that of a reference flow of carrier gas alone is measured. In general, the thermal conductivity detector responds uniformly to volatile compounds regardless of structure; however, it is considerably less sensitive than the flame-ionization detector.
The alkali flame-ionization detector, sometimes called an NP or nitrogen-phosphorus detector, contains a thermionic source, such as an alkali-metal salt or a glass element containing rubidium or other metal, that results in the efficient ionization of organic nitrogen and phosphorus compounds. It is a selective detector that shows little response to hydrocarbons.
The electron-capture detector contains a radioactive source of ionizing radiation. It exhibits an extremely high response to compounds containing halogens and nitro groups but little response to hydrocarbons. The sensitivity increases with the number and atomic weight of the halogen atoms.
Data Collection Devices—Modern data stations receive the detector output, calculate peak areas and peak heights, and print chromatograms, complete with run parameters and peak data. Chromatographic data may be stored and reprocessed, with integration and other calculation variables being changed as required. Data stations are used also to program the chromatograph, controlling most operational variables and providing for long periods of unattended operation.
Data can also be collected for manual measurement on simple recorders or on integrators whose capabilities range from those providing a printout of peak areas to those providing chromatograms with peak areas and peak heights calculated and data stored for possible reprocessing.
Procedure—
Packed and capillary columns must be conditioned before use until the baseline and other characteristics are stable. This may be done by operation at a temperature above that called for by the method or by repeated injections of the compound or mixture to be chromatographed. The column or packing material supplier provides instructions for the recommended conditioning procedure. In the case of thermally stable methyl- and phenyl-substituted polysiloxanes, a special sequence increases inertness and efficiency; maintain the column at a temperature of 250 for 1 hour, with helium flowing, to remove oxygen and solvents. Stop the flow of helium, heat at about 340 for 4 hours, then reduce the heating to a temperature of 250, and condition with helium flowing until stable.
Most drugs are reactive polar molecules. Successful chromatography may require conversion of the drug to a less polar and more volatile derivative by treatment of reactive groups with appropriate reagents. Silylating agents are widely used for this purpose and are readily available.
Assays require quantitative comparison of one chromatogram with another. A major source of error is irreproducibility in the amount of sample injected, notably when manual injections are made with a syringe. The effects of variability can be minimized by addition of an internal standard, a noninterfering compound present at the same concentration in test and standard solutions. The ratio of peak response of the analyte to that of the internal standard is compared from one chromatogram to another. Where the internal standard is chemically similar to the substance being determined, there is also compensation for minor variations in column and detector characteristics. In some cases, the internal standard may be carried through the sample preparation procedure prior to gas chromatography to control other quantitative aspects of the assay. Automatic injectors greatly improve the reproducibility of sample injections and reduce the need for internal standards.
Many monographs require that system suitability requirements be met before samples are analyzed (see System Suitability and Interpretation of Chromatograms).
HIGH-PRESSURE LIQUID CHROMATOGRAPHY
High-pressure liquid chromatography (HPLC), sometimes called high-performance liquid chromatography, is a separation technique based on a solid stationary phase and a liquid mobile phase. Separations are achieved by partition, adsorption, or ion-exchange processes, depending upon the type of stationary phase used. HPLC has distinct advantages over gas chromatography for the analysis of organic compounds. Compounds to be analyzed are dissolved in a suitable solvent, and most separations take place at room temperature. Thus, most drugs, being nonvolatile or thermally unstable compounds, can be chromatographed without decomposition or the necessity of making volatile derivatives. Most pharmaceutical analyses are based on partition chromatography and are completed within 30 minutes.
As in gas chromatography, the elution time of a compound can be described by the capacity factor, k¢ (see Glossary of Symbols), which depends on the chemical nature of the analyte, the composition and flow rate of the mobile phase, and the composition and surface area of the stationary phase. Column length is an important determinant of resolution. Only compounds having different capacity factors can be separated by HPLC.
Apparatus—
A liquid chromatograph consists of a reservoir containing the mobile phase, a pump to force the mobile phase through the system at high pressure, an injector to introduce the sample into the mobile phase, a chromatographic column, a detector, and a data collection device such as a computer, integrator, or recorder. Short, small-bore columns containing densely packed particles of stationary phase provide for the rapid exchange of compounds between the mobile and stationary phases. In addition to receiving and reporting detector output, computers are used to control chromatographic settings and operations, thus providing for long periods of unattended operation.
Pumping Systems—HPLC pumping systems deliver metered amounts of mobile phase from the solvent reservoirs to the column through high-pressure tubing and fittings. Modern systems consist of one or more computer-controlled metering pumps that can be programmed to vary the ratio of mobile phase components, as is required for gradient chromatography, or to mix isocratic mobile phases (i.e., mobile phases having a fixed ratio of solvents). However, the proportion of ingredients in premixed isocratic mobile phases can be more accurately controlled than in those delivered by most pumping systems. Operating pressures up to 5000 psi or higher, with delivery rates up to about 10 mL per minute are typical. Pumps used for quantitative analysis should be constructed of materials inert to corrosive mobile phase components and be capable of delivering the mobile phase at a constant rate with minimal fluctuations over extended periods of time.
Injectors—After dissolution in mobile phase or other suitable solution, compounds to be chromatographed are injected into the mobile phase, either manually by syringe or loop injectors, or automatically by autosamplers. The latter consist of a carousel or rack to hold sample vials with tops that have a pierceable septum or stopper and an injection device to transfer sample from the vials to a loop from which it is loaded into the chromatograph. Some autosamplers can be programmed to control sample volume, the number of injections and loop rinse cycles, the interval between injections, and other operating variables.
A syringe can be used for manual injection of samples through a septum when column head pressures are less than 70 atmospheres (about 1000 psi). At higher pressures an injection valve is essential. Some valve systems incorporate a calibrated loop that is filled with test solution for transfer to the column in the mobile phase. In other systems, the test solution is transferred to a cavity by syringe and then switched into the mobile phase.
Columns—For most pharmaceutical analyses, separation is achieved by partition of compounds in the test solution between the mobile and stationary phases. Systems consisting of polar stationary phases and nonpolar mobile phases are described as normal phase, while the opposite arrangement, polar mobile phases and nonpolar stationary phases, is called reverse-phase chromatography. Partition chromatography is almost always used for hydrocarbon-soluble compounds of molecular weight less than 1000. The affinity of a compound for the stationary phase, and thus its retention time on the column, is controlled by making the mobile phase more or less polar. Mobile phase polarity can be varied by the addition of a second, and sometimes a third or even a fourth, component.
Stationary phases for modern, reverse-phase liquid chromatography typically consist of an organic phase chemically bound to silica or other materials. Particles are usually 3 to 10 µm in diameter, but sizes may range up to 50 µm or more for preparative columns. Small particles thinly coated with organic phase provide for low mass transfer resistance and, hence, rapid transfer of compounds between the stationary and mobile phases. Column polarity depends on the polarity of the bound functional groups, which range from relatively nonpolar octadecyl silane to very polar nitrile groups. Liquid, nonbound stationary phases must be largely immiscible in the mobile phase. Even so, it is usually necessary to presaturate the mobile phase with stationary phase to prevent stripping of the stationary phase from the column. Polymeric stationary phases coated on the support are more durable.
Columns used for analytical separations usually have internal diameters of 2 to 5 mm; larger diameter columns are used for preparative chromatography. Columns may be heated to give more efficient separations, but only rarely are they used at temperatures above 60 because of potential stationary phase degradation or mobile phase volatility. Unless otherwise specified in the individual monograph, columns are used at ambient temperature.
Ion-exchange chromatography is used to separate water-soluble, ionizable compounds of molecular weight less than 1500. The stationary phases are usually synthetic organic resins; cation-exchange resins contain negatively charged active sites and are used to separate basic substances such as amines, while anion-exchange resins have positively charged active sites for separation of compounds with negatively charged groups, such as phosphate, sulfonate, or carboxylate groups. Water-soluble ionic or ionizable compounds are attracted to the resins, and differences in affinity bring about the chromatographic separation. The pH of the mobile phase, temperature, ion type, ionic concentration, and organic modifiers affect the equilibrium, and these variables can be adjusted to obtain the desired degree of separation.
In size-exclusion chromatography, columns are packed with a porous stationary phase. Molecules of the compounds being chromatographed are filtered according to size. Those too large to enter the pores pass unretained through the column. Smaller molecules enter the pores and are increasingly retained as molecular size decreases. These columns are typically used to measure aggregation and degradation of large molecules (see Size-Exclusion Chromatography section).
Detectors—Many compendial HPLC methods require the use of spectrophotometric detectors. Such a detector consists of a flow-through cell mounted at the end of the column. A beam of UV radiation passes through the flow cell and into the detector. As compounds elute from the column, they pass through the cell and absorb the radiation, resulting in measurable energy level changes.
Fixed, variable, and multi-wavelength detectors are widely available. Fixed wavelength detectors operate at a single wavelength, typically 254 nm, emitted by a low-pressure mercury lamp. Variable wavelength detectors contain a continuous source, such as a deuterium or high-pressure xenon lamp, and a monochromator or an interference filter to generate monochromatic radiation at a wavelength selected by the operator. The wavelength accuracy of a variable-wavelength detector equipped with a monochromator should be checked by the procedure recommended by its manufacturer; if the observed wavelengths differ by more than 3 nm from the correct values, recalibration of the instrument is indicated. Modern variable wavelength detectors can be programmed to change wavelength while an analysis is in progress. Multi-wavelength detectors measure absorbance at two or more wavelengths simultaneously. In diode array multi-wavelength detectors, continuous radiation is passed through the sample cell, then resolved into its constituent wavelengths, which are individually detected by the photodiode array. These detectors acquire absorbance data over the entire UV-visible range, thus providing the analyst with chromatograms at multiple, selectable wavelengths and spectra of the eluting peaks. Diode array detectors usually have lower signal-to-noise ratios than fixed or variable wavelength detectors, and thus are less suitable for analysis of compounds present at low concentrations.
Differential refractometer detectors measure the difference between the refractive index of the mobile phase alone and that of the mobile phase containing chromatographed compounds as it emerges from the column. Refractive index detectors are used to detect non-UV absorbing compounds, but they are less sensitive than UV detectors. They are sensitive to small changes in solvent composition, flow rate, and temperature, so that a reference column may be required to obtain a satisfactory baseline.
Fluorometric detectors are sensitive to compounds that are inherently fluorescent or that can be converted to fluorescent derivatives either by chemical transformation of the compound or by coupling with fluorescent reagents at specific functional groups. If derivatization is required, it can be done prior to chromatographic separation or, alternatively, the reagent can be introduced into the mobile phase just prior to its entering the detector.
Potentiometric, voltametric, or polarographic electrochemical detectors are useful for the quantitation of species that can be oxidized or reduced at a working electrode. These detectors are selective, sensitive, and reliable, but require conducting mobile phases free of dissolved oxygen and reducible metal ions. A pulseless pump must be used, and care must be taken to ensure that the pH, ionic strength, and temperature of the mobile phase remain constant. Working electrodes are prone to contamination by reaction products with consequent variable responses.
Electrochemical detectors with carbon-paste electrodes may be used advantageously to measure nanogram quantities of easily oxidized compounds, notably phenols and catechols.
New detectors continue to be developed in attempts to overcome the deficiencies of those being used.
Data Collection Devices—Modern data stations receive and store detector output and print out chromatograms complete with peak heights, peak areas, sample identification, and method variables. They are also used to program the liquid chromatograph, controlling most variables and providing for long periods of unattended operation.
Data also may be collected on simple recorders for manual measurement or on stand-alone integrators, which range in complexity from those providing a printout of peak areas to those providing chromatograms with peak areas and peak heights calculated and data stored for possible subsequent reprocessing.
Procedure—
The mobile phase composition significantly influences chromatographic performance and the resolution of compounds in the mixture being chromatographed. For accurate quantitative work, high-purity reagents and “HPLC grade” organic solvents must be used. Water of suitable quality should have low conductivity and low UV absorption, appropriate to the intended use.
Reagents used with special types of detectors (e.g., electrochemical, mass spectrometer) may require the establishment of additional tolerances for potential interfering species. Composition has a much greater effect than temperature on the capacity factor, k¢.
In partition chromatography, the partition coefficient, and hence the separation, can be changed by addition of another component to the mobile phase. In ion-exchange chromatography, pH and ionic strength, as well as changes in the composition of the mobile phase, affect capacity factors. The technique of continuously changing the solvent composition during the chromatographic run is called gradient elution or solvent programming. It is sometimes used to chromatograph complex mixtures of components differing greatly in their capacity factors. Detectors that are sensitive to change in solvent composition, such as the differential refractometer, are more difficult to use with the gradient elution technique.
The detector must have a broad linear dynamic range, and compounds to be measured must be resolved from any interfering substances. The linear dynamic range of a compound is the range over which the detector signal response is directly proportional to the amount of the compound. For maximum flexibility in quantitative work, this range should be about three orders of magnitude. HPLC systems are calibrated by plotting peak responses in comparison with known concentrations of a reference standard, using either an external or an internal standardization procedure.
Reliable quantitative results are obtained by external calibration if automatic injectors or autosamplers are used. This method involves direct comparison of the peak responses obtained by separately chromatographing the test and reference standard solutions. If syringe injection, which is irreproducible at the high pressures involved, must be used, better quantitative results are obtained by the internal calibration procedure where a known amount of a noninterfering compound, the internal standard, is added to the test and reference standard solutions, and the ratios of peak responses of drug and internal standard are compared.
Because of normal variations in equipment, supplies, and techniques, a system suitability test is required to ensure that a given operating system may be generally applicable. The main features of system suitability tests are described below.
For information on the interpretation of results, see the section Interpretation of Chromatograms.
Size-Exclusion Chromatography
Size-exclusion chromatography is a high-pressure liquid chromatographic technique that separates molecules in solution according to their size. Methods for size-exclusion chromatography are divided into gel permeation chromatographic methods, which utilize nonpolar organic mobile phases and hydrophilic packings, and gel filtration chromatographic methods, which utilize aqueous mobile phases and hydrophobic packings. The sample is introduced into a column, which is filled with a gel or a porous particle packing material and is carried by the mobile phase through the column. The size separation takes place by repeated exchange of the solute molecules between the solvent of the mobile phase and the same solvent in the stationary liquid phase within the pores of the packing material. The pore-size range of the packing material determines the molecular-size range within which separation can occur.
Molecules small enough to penetrate all the pore spaces elute at the total permeation volume, VT. On the other hand, molecules apparently larger than the maximum pore size of the packing material migrate along the column only through the spaces between the particles of the packing material without being retained and elute at the exclusion volume, VO (void volume). Separation according to molecular size occurs between the exclusion volume and the total permeation volume, useful separation usually occurring in the first two-thirds of this range.
Apparatus—
The components of the chromatograph are described under High-Pressure Liquid Chromatography.
Column—If necessary, the column is temperature-controlled. It is packed with a separation material that is capable of fractionation in the appropriate range of molecular sizes and through which the eluant is passed at a constant rate. One end of the column is usually fitted with a suitable device for applying the sample, such as a flow adaptor, a syringe through a septum or an injection valve, and it may also be connected to a suitable pump for controlling the flow of the eluant. Alternatively, the sample may be applied directly to the drained bed surface, or, where the sample is denser than the eluant, it may be layered beneath the eluant. The packing material may be a soft support such as a swollen gel or a rigid support composed of a material such as glass, silica, or a solvent-compatible, cross-linked organic polymer. Rigid supports usually require pressurized systems giving faster separations. The mobile phase is chosen according to sample type, separation medium, and method of detection.
Detector—The outlet of the column is usually connected to a suitable detector fitted with an automatic recorder that enables the monitoring of the relative concentrations of separated components of the sample. Detectors are usually based on photometric, refractometric, or luminescent properties (see Detectors under High-Pressure Liquid Chromatography). An automatic fraction collector may be attached, if necessary.
Procedure—
Before carrying out the separation, the packing material is treated and the column is packed, as described in the individual monograph or according to the manufacturer's instructions. Where necessary, procedures for verifying the suitability of the system are described in the individual monograph. The column efficiency may be evaluated from the number of theoretical plates, N (see the section Interpretation of Chromatograms). The elution characteristics of a compound in a particular column may be described by the distribution coefficient, KD, which is calculated by the formula:
(VI – VO) / (VT – VO)
in which VO, V T, and VI are the retention volumes for the nonretained component, the component that has full access to all the pores in the support, and the compound under test, respectively. Each retention volume is measured from the time of application to the time of the peak maximum.
Determination of Relative Component Composition of Mixture—Carry out the separation as directed in the individual monograph. Monitor the elution of the components continuously, and measure the corresponding peak areas. If all the components under test exhibit equivalent responses to the physicochemical property being monitored (for example, if they exhibit corresponding absorptivities), calculate the relative amount of each component by dividing the respective peak area by the sum of the peak areas of all the components under test. If the responses to the property used for detection of the components under test are not equivalent, calculate the content using calibration curves obtained from the calibration procedure specified in the individual monograph.
Determination of Molecular Weights—Size-exclusion chromatography is used to determine molecular weights of components under test by comparison to calibration standards specified in the individual monograph. Plot the retention volumes of the calibration standards versus the logarithm of their molecular weights. Draw the line that best fits the plotted points within the exclusion and total permeation limits for the particular separation medium. From the calibration curve, molecular weights of components under test are estimated. This calibration is valid only for the particular macromolecular solute-solvent system used under the specified experimental conditions.
Determination of Molecular Weight Distribution of Polymers—The material used for calibration and the methods for determination of the distribution of molecular weights of polymers are specified in the individual monograph. However, sample comparison is valid only for results obtained under identical experimental conditions.
INTERPRETATION OF CHROMATOGRAMS
Figure 1 represents a typical chromatographic separation of two substances, 1 and 2, where t1 and t2 are the respective retention times; and h, h/2, and Wh/2 are the height, the half-height, and the width at half-height, respectively, for peak 1. W1 and W2 are the respective widths of peaks 1 and 2 at the baseline. Air peaks are a feature of gas chromatograms and correspond to the solvent front in liquid chromatography. The retention time of these unretained components is designated as tM.
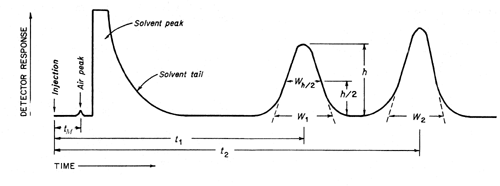
Figure 1. Chromatographic separation of two substances
Chromatographic separation of two substances
Chromatographic retention times are characteristic of the compounds they represent but are not unique. Coincidence of retention times of a test and a reference substance can be used as a feature in construction of an identity profile but is insufficient on its own to establish identity. Absolute retention times of a given compound vary from one chromatogram to the next.
Because in most procedures there is no need to identify an unretained peak, comparisons are normally made in terms of relative retention times, Rr:
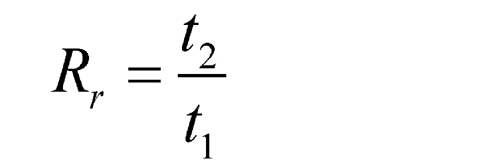
where t2 and t1 are the retention times, measured from the point of injection, of the test and the reference substances, respectively, determined under identical experimental conditions on the same column.
Other procedures may identify the peak position using the relative retention, r:
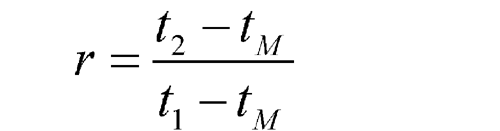
where tM is the retention time of a non-retained marker, which needs to be defined in the procedure.
The number of theoretical plates, N, is a measure of column efficiency. For Gaussian peaks, it is calculated by the equation:
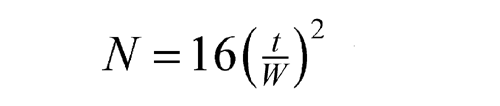
where t is the retention time of the substance and W is the width of the peak at its base, obtained by extrapolating the relatively straight sides of the peak to the baseline. The value of N depends upon the substance being chromatographed as well as the operating conditions such as mobile phase or carrier gas flow rates and temperature, the quality of the packing, the uniformity of the packing within the column and, for capillary columns, the thickness of the stationary phase film, and the internal diameter and length of the column.
The separation of two components in a mixture, the resolution, R, is determined by the equation:
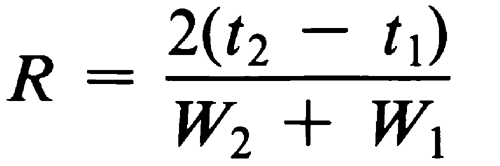
in which t2 and t1 are the retention times of the two components, and W2 and W1 are the corresponding widths at the bases of the peaks obtained by extrapolating the relatively straight sides of the peaks to the baseline.
Where electronic integrators are used, it may be convenient to determine the resolution, R, by the equation:
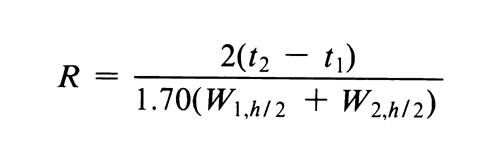
and to determine the number of theoretical plates, N, by the equation:
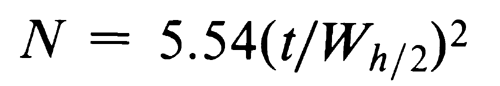
where Wh/2 is the peak width at half-height, obtained directly by electronic integrators. However, in the event of dispute, only equations based on peak width at baseline are to be used.
Peak areas and peak heights are usually proportional to the quantity of compound eluting. These are commonly measured by electronic integrators but may be determined by more classical approaches. Peak areas are generally used but may be less accurate if peak interference occurs. For manual measurements, the chart should be run faster than usual, or a comparator should be used to measure the width at half-height and the width at the base of the peak, to minimize error in these measurements. For accurate quantitative work, the components to be measured should be separated from any interfering components. Peak tailing and fronting and the measurement of peaks on solvent tails are to be avoided.
Chromatographic purity tests for drug raw materials are sometimes based on the determination of peaks due to impurities, expressed as a percentage of the area due to the drug peak. It is preferable, however, to compare impurity peaks with those in the chromatogram of a standard at a similar concentration. The standard may be the drug itself at a level corresponding to, for example, 0.5% impurity, or in the case of toxic or signal impurities, a standard of the impurity itself.
SYSTEM SUITABILITY
System suitability tests are an integral part of gas and liquid chromatographic methods. They are used to verify that the resolution and reproducibility of the chromatographic system are adequate for the analysis to be done. The tests are based on the concept that the equipment, electronics, analytical operations, and samples to be analyzed constitute an integral system that can be evaluated as such.
The resolution, R, [note—All terms and symbols are defined in the Glossary of Symbols] is a function of column efficiency, N, and is specified to ensure that closely eluting compounds are resolved from each other, to establish the general resolving power of the system, and to ensure that internal standards are resolved from the drug. Column efficiency may be specified also as a system suitability requirement, especially if there is only one peak of interest in the chromatogram; however, it is a less reliable means to ensure resolution than direct measurement. Column efficiency is a measure of peak sharpness, which is important for the detection of trace components.
Replicate injections of a standard preparation used in the assay or other standard solution are compared to ascertain whether requirements for precision are met. Unless otherwise specified in the individual monograph, data from five replicate injections of the analyte are used to calculate the relative standard deviation, SR, if the requirement is 2.0% or less; data from six replicate injections are used if the relative standard deviation requirement is more than 2.0%.
The tailing factor, T, a measure of peak symmetry, is unity for perfectly symmetrical peaks and its value increases as tailing becomes more pronounced (see Figure 2). In some cases, values less than unity may be observed. As peak asymmetry increases, integration, and hence precision, becomes less reliable.
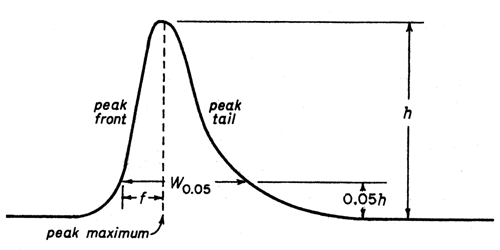
Figure 2.Asymmetrical chromatographic peak
These tests are performed by collecting data from replicate injections of standard or other solutions as specified in the individual monograph. The specification of definitive parameters in a monograph does not preclude the use of other suitable operating conditions (see Procedures under Tests and Assays in the General Notices). If adjustments of operating conditions to meet system suitability requirements are necessary, each of the following is the maximum variation that can be considered, unless otherwise directed in the monograph. Adjustments are permitted only when suitable standards (including Reference Standards) are available for all compounds used in the suitability test and only when those standards are used to show that the adjustments have improved the quality of the chromatography in meeting system suitability requirements. Adjustments to chromatographic systems performed in order to comply with system suitability requirements are not to be made to compensate for column failure or system malfunction. The changes described below may require additional validation data. The user should verify the suitability of the method under the new conditions by assessing the relevant analytical performance characteristics potentially affected by the change. Multiple adjustments can have a cumulative effect in the performance of the system and should be considered carefully before implementation.
pH of Mobile Phase (HPLC)—
The pH of the aqueous buffer used in the preparation of the mobile phase can be adjusted to within ±0.2 units of the value or range specified.
Concentration of Salts in Buffer (HPLC)—
The concentration of the salts used in the preparation of the aqueous buffer used in the mobile phase can be adjusted to within ±10%, provided the permitted pH variation (see above) is met.
Ratio of Components in Mobile Phase (HPLC)—
The following adjustment limits apply to minor components of the mobile phase (specified at 50% or less). The amount(s) of these component(s) can be adjusted by ±30% relative. However, the change in any component cannot exceed ±10% absolute (i.e., in relation to the total mobile phase). Adjustment can be made to one minor component in a ternary mixture. Examples of adjustments for binary and ternary mixtures are given below.
Binary Mixtures—
specified ratio of 50:50
—Thirty percent of 50 is 15% absolute, but this exceeds the maximum permitted change of ±10% absolute in either component. Therefore, the mobile phase ratio may be adjusted only within the range of 40:60 to 60:40.
specified ratio of 2:98
—Thirty percent of 2 is 0.6% absolute. Therefore the maximum allowed adjustment is within the range of 1.4:98.6 to 2.6:97.4.
Ternary Mixtures—
specified ratio of 60:35:5
—For the second component, 30% of 35 is 10.5% absolute, which exceeds the maximum permitted change of ±10% absolute in any component. Therefore the second component may be adjusted only within the range of 25% to 45% absolute. For the third component, 30% of 5 is 1.5% absolute. In all cases, a sufficient quantity of the first component is used to give a total of 100%. Therefore, mixture ranges of 50:45:5 to 70:25:5 or 58.5:35:6.5 to 61.5:35:3.5 would meet the requirement.
Wavelength of UV-Visible Detector (HPLC)—
Deviations from the wavelengths specified in the method are not permitted. The procedure specified by the detector manufacturer, or another validated procedure, is to be used to verify that error in the detector wavelength is, at most, ±3 nm.
Column Length (GC, HPLC):
can be adjusted by as much as ±70%.
Column Inner Diameter (GC, HPLC):
can be adjusted by as much as ±25% for HPLC and ±50% for GC.
Film Thickness (Capillary GC):
can be adjusted by as much as –50% to 100%.
Particle Size (HPLC):
can be reduced by as much as 50%.
Particle Size (GC):
going from a larger to a smaller or a smaller to a larger (if it is the same “Range Ratio”, which is the diameter of the largest particle divided by the diameter of the smallest particle) particle size GC mesh support is acceptable, provided the chromatography meets the requirements of the system suitability.
Flow Rate (GC, HPLC):
can be adjusted by as much as ±50%.
Injection Volume (GC, HPLC):
can be reduced as far as is consistent with accepted precision and detection limits.
Column Temperature (HPLC):
can be adjusted by as much as ±10. Column thermostating is recommended to improve control and reproducibility of retention time.
Oven Temperature (GC):
can be adjusted by as much as ±10%.
Oven Temperature Program (GC)—
Adjustment of temperatures is permitted as stated above. For the times specified for the temperature to be maintained or for the temperature to be changed from one value to another, an adjustment of up to ±20% is permitted.
Unless otherwise directed in the monograph, system suitability parameters are determined from the analyte peak.
Relative retention times may be provided in monographs for informational purposes only, to aid in peak identification. There are no acceptance criteria applied to relative retention times.
To ascertain the effectiveness of the final operating system, it should be subjected to suitability testing. Replicate injections of the standard preparation required to demonstrate adequate system precision may be made before the injection of samples or may be interspersed among sample injections. System suitability must be demonstrated throughout the run by injection of an appropriate control preparation at appropriate intervals, including at the end of the analysis. The control preparation can be a standard preparation or a solution containing a known amount of analyte and any additional materials useful in the control of the analytical system, such as excipients or impurities. Whenever there is a significant change in equipment or in a critical reagent, suitability testing should be performed before the injection of samples. No sample analysis is acceptable unless the requirements of system suitability have been met. Sample analyses obtained while the system fails system suitability requirements are unacceptable.
Change to read:
GLOSSARY OF SYMBOLS
To promote uniformity of interpretation, the following symbols and definitions are employed where applicable in presenting formulas in the individual monographs. Where a different symbol or definition is used in an individual monograph, the monograph text takes precedence (see General Notices). [note—Where the terms W and t both appear in the same equation they must be expressed in the same units.]
| f | distance from the peak maximum to the leading edge of the peak, the distance being measured at a point 5% of the peak height from the baseline. | |
| k¢ | capacity factor, |
|
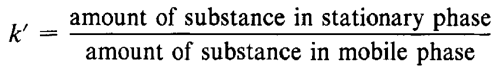
|
||

|
||
| N | number of theoretical plates in a chromatographic column, |
|
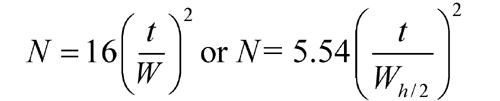
|
||
| r | relative retention, |
|
|
|
||
| ri | peak response of an impurity obtained from a chromatogram. | |
| rIS | peak response of the internal standard obtained from a chromatogram. | |
| rS | peak response of the Reference Standard obtained from a chromatogram. | |
| rU | peak response of the analyte obtained from a chromatogram. | |
| R | resolution between two chromatographic peaks, | |
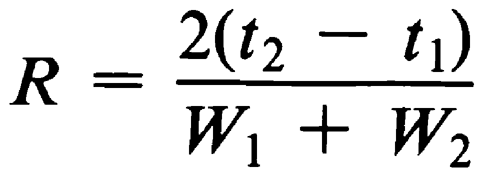
|
||
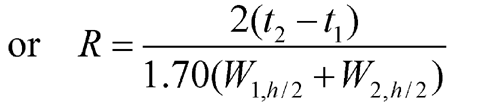
|
||
| RF | chromatographic retardation factor equal to the ratio of the distance from the origin to the center of a zone divided by the distance from the origin to the solvent front. | |
| Rr | relative retention time, |
|
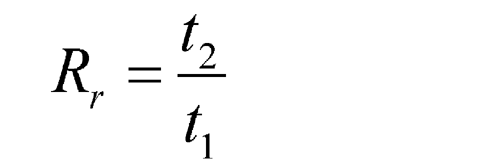
|
||
| Rrel | relative retardation | |
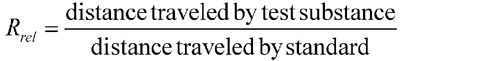
|
||
| RS | peak response ratio for a Standard preparation containing Reference Standard and internal standard, | |
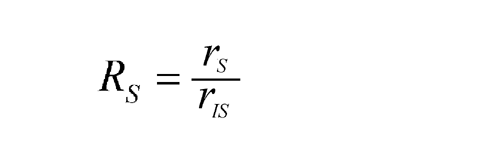
|
||
| RU | peak response ratio for Assay preparation containing the analyte and internal standard, | |
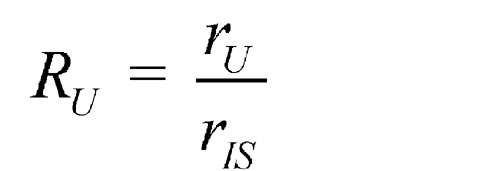
|
||
| percent relative standard deviation, | ||
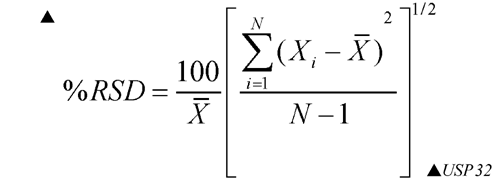
|
||
| where Xi is an individual measurement in a set of N measurements and X is the arithmetic mean of the set. | ||
| T | tailing factor, |
|
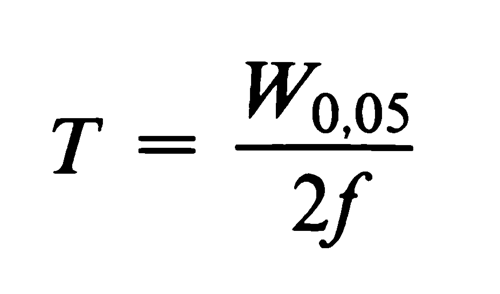
|
||
| t | retention time measured from time of injection to time of elution of peak maximum. | |
| tM | retention time of nonretarded component, air with thermal conductivity detection. | |
| W | width of peak measured by extrapolating the relatively straight sides to the baseline. | |
| Wh/2 | width of peak at half height. | |
| W0.05 | width of peak at 5% height. | |
|
|
||
|
|
||
CHROMATOGRAPHIC REAGENTS
A complete list of Packings (L), Phases (G), and Supports (S) used in USP–NF tests and assays is located under Chromatographic Reagents in the Reagents, Indicators, and Solutions section. This list is intended to be a convenient reference for the chromatographer to identify the pertinent chromatographic reagent specified in the individual monograph.
[note—Particle sizes given in the listing are those generally provided. Where other, usually finer, sizes are required, the individual monograph specifies the desired particle size. Within any category of packings or phases listed there may be a wide range of columns available. Where it is necessary to define more specifically the chromatographic conditions, the individual monograph so indicates.]
1
A suitable grade is acid-washed Celite 545, available from Johns-Manville Corp., 22 East 40th St., New York, NY 10016.
Auxiliary Information— Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
| General Chapter | Horacio N. Pappa, Ph.D.
Senior Scientist and Latin American Liaison 1-301-816-8319 |
(GC05) General Chapters 05 |
USP32–NF27 Page 227
Pharmacopeial Forum: Volume No. 34(5) Page 1238