Pravastatin Sodium Tablets
» Pravastatin Sodium contains not less than 90.0 percent and not more than 110.0 percent of the labeled amount of pravastatin sodium (C23H35NaO7).
Packaging and storage—
Preserve in tight containers. Protect from moisture and light. Store at controlled room temperature.
USP Reference standards  11
11 —
—
USP Pravastatin Related Compound B RS .
.
USP Pravastatin Sodium RS .
USP Pravastatin 1,1,3,3-Tetramethylbutylamine RS.
USP Pravastatin Related Compound B RS
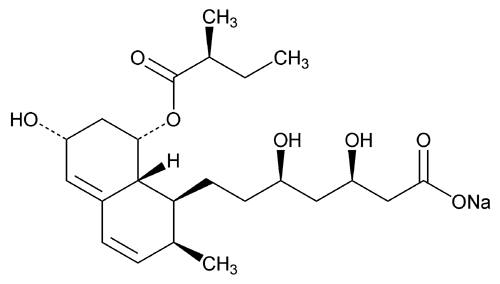 .
.
USP Pravastatin Sodium RS
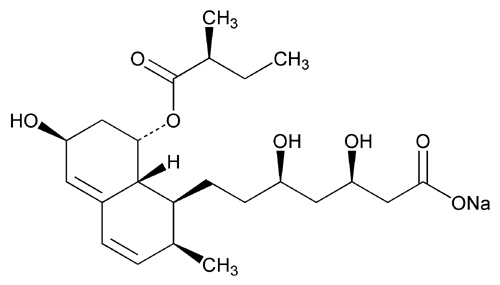 .
.
USP Pravastatin 1,1,3,3-Tetramethylbutylamine RS.
Identification—
A:
The retention time of the major peak in the chromatogram of the Assay preparation corresponds to that in the chromatogram of the Standard preparation, as obtained in the Assay.
B:
Ultraviolet Absorption 197U—Finely powder a number of Tablets, and extract with water a portion equivalent to about 10 mg of pravastatin sodium. The UV absorption spectrum of a solution of pravastatin sodium in water containing about 10 µg per mL exhibits maxima at the same wavelength as that of a similar solution of USP Pravastatin Sodium RS, concomitantly measured between 220 and 340 nm.
Dissolution 711—
Medium:
water; 900 mL.
Apparatus 2:
50 rpm.
Time:
30 minutes.
Procedure—
Determine the amount of C23H35NaO7 dissolved by employing UV absorption at the wavelength of maximum absorbance at about 238 nm on filtered portions of the solution under test, suitably diluted with Medium, if necessary, in comparison with a Standard solution having a known concentration of USP Pravastatin 1,1,3,3-Tetramethylbutylamine RS in the same Medium. [note—To express the concentration of the Standard solution as pravastatin sodium, use the conversion factor of (446.51/553.78), in which 446.51 and 553.78 are the molecular weights of pravastatin sodium and pravastatin 1,1,3,3-tetramethylbutylamine, respectively.]
Tolerances—
Not less than 80% (Q) of the labeled amount of C23H35NaO7 is dissolved in 30 minutes.
Uniformity of dosage units 905:
meet the requirements.
Related compounds—
Mobile phase and Chromatographic system—
Proceed as directed in the Assay.
Test solution—
Use the Assay preparation, prepared as directed in the Assay. [note—Use this solution within 24 hours of preparation.]
Procedure—
Inject a volume (about 20 µL) of the Test solution into the chromatograph, record the chromatograms for up to 4 times the retention time of the pravastatin peak, identify the impurities listed in Table 1, and measure the peak responses. Calculate the percentage of each impurity in the portion of Tablets taken by the formula:
100(ri / rs)
in which ri is the peak response of the individual impurity, and rs is the sum of the responses of all the peaks obtained from the Test solution. In addition to not exceeding the limits of each impurity in Table 1, not more than 0.2% of any unspecified individual impurity is found, and not more than 3% of total impurities is found. Disregard the peak due to pravastatin related compound B that elutes at the relative retention time of about 0.7 and the peak due to 3¢¢-hydroxypravastatin at the relative retention time of about 0.3, as these impurities are controlled in the drug substance monograph. Disregard any impurity that is less than 0.05%.
Table 1
| Name | Relative Retention Time |
Limit (%) |
| Oxidation impurity1 | 0.5 | 1 |
| Pravastatin sodium | 1.0 | n/a |
| Specified unknown impurity 1 | 1.6 | 0.2 |
| Specified unknown impurity 2 | 1.8 | 0.2 |
| Pravastatin lactone | 2.1 | 2 |
| Specified unknown impurity 3 | 2.8 | 0.2 |
| Specified unknown impurity 4 | 3.2 | 0.2 |
| Specified unknown impurity 5 | 3.8 | 0.2 |
|
1
Sodium (3R,5R)-3,5-dihydroxy-7-((1S,2S)-6-hydroxy-2-methyl-1,2-dihydronaphthalen-1-yl)heptanoate.
|
||
Assay—
[note—The Standard preparation, Assay stock preparation, and Assay preparation can be stored for up to 7 days at room temperature.]
Mobile phase—
Prepare a filtered and degassed mixture of methanol, water, glacial acetic acid, and triethylamine (500:500:1:1). Make adjustments if necessary (see System Suitability under Chromatography 621).
Diluent 1—
Transfer 16.4 g of anhydrous sodium acetate into a 2000-mL volumetric flask. Add 1600 mL of water, adjust with glacial acetic acid to a pH of 5.6, dilute with water to volume, and mix.
Diluent 2—
Prepare a mixture of Diluent 1 and methanol (80:20).
Standard preparation—
Transfer an accurately weighed quantity of USP Pravastatin 1,1,3,3-Tetramethylbutylamine RS to a suitable volumetric flask, and dissolve in Diluent 1 using sonication to obtain a solution having a known concentration of about 0.6 mg of pravastatin 1,1,3,3-tetramethylbutylamine per mL. Dilute 5.0 mL of this solution with Diluent 2 to 25.0 mL, and mix.
Assay stock preparation—
Transfer not fewer than 5 Tablets to a suitable volumetric flask with at least a (NL×2)-mL capacity, N being the number of Tablets transferred, and L being the label claim per Tablet, filled to at least 80% capacity with Diluent 1. [note—It is necessary to fill the flask to 80% capacity to maintain the correct pH throughout the preparation.] Shake for at least 1 hour, and sonicate for at least 15 minutes with periodic shaking of the flask by hand, until the Tablets have completely disintegrated. Allow to cool, and dilute with Diluent 1 to volume. Centrifuge a portion of the solution for 15 minutes at 2000 rpm, or filter.
Assay preparation—
Dilute approximately 5 mL of the Assay stock preparation with Diluent 2 to obtain a solution having an expected concentration of about 0.1 mg per mL, based on the label claim.
Resolution solution—
Transfer about 2 mg of USP Pravastatin Related Compound B RS to a 10-mL volumetric flask. Dissolve in and dilute with methanol to volume. Transfer 0.1 mL of this solution and 1.0 mL of the Standard preparation to a small tube, and mix. [note—Pravastatin related compound B is the 6¢-epipravastatin sodium.]
Chromatographic system (see Chromatography 621)—
The liquid chromatograph is equipped with a 238-nm detector and a 4.6-mm × 5-cm column than contains endcapped packing L1. Alternatively, a 3.9-mm × 7.5-cm column containing endcapped packing L1 can be used. The flow rate is about 1.0 mL per minute. Chromatograph the Resolution solution, and record the peak responses as directed for Procedure: the relative retention times are about 0.7 for pravastatin related compound B and 1.0 for pravastatin; the resolution, R, between the pravastatin related compound B and the pravastatin peaks is not less than 3.0. Chromatograph the Standard preparation, and record the peak responses as directed for Procedure: the relative standard deviation for replicate injections is not more than 2.0%.
Procedure—
Separately inject equal volumes (about 20 µL) of the Standard preparation and the Assay preparation into the chromatograph, record the chromatograms, and measure the peak response for pravastatin. Calculate the percentage of pravastatin sodium (C23H35NaO7) in the portion of Tablets taken by the formula:
100(446.51/553.78)(CVD/NL)(rU / rS)
in which 100 is the conversion factor to percentage; 446.51 and 553.78 are the molecular weights of pravastatin sodium and pravastatin 1,1,3,3-tetramethylbutylamine, respectively; C is the concentration, in mg per mL, of pravastatin 1,1,3,3-tetramethylbutylamine in the Standard preparation; V is the volume, in mL, of the Assay stock preparation; D is the dilution factor of the Assay preparation; N is the number of Tablets taken to prepare the Assay stock preparation; L is the label claim, in mg of pravastatin sodium per Tablet; and rU and rS are the pravastatin peak responses obtained from the Assay preparation and the Standard preparation, respectively.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
Chromatographic Column—
| Topic/Question | Contact | Expert Committee |
| Monograph | Elena Gonikberg, Ph.D.
Senior Scientist 1-301-816-8251 |
(MDGRE05) Monograph Development-Gastrointestinal Renal and Endocrine |
| Reference Standards | Lili Wang, Technical Services Scientist 1-301-816-8129 RSTech@usp.org |
|
| Margareth R.C. Marques, Ph.D.
Senior Scientist 1-301-816-8106 |
(BPC05) Biopharmaceutics05 |
USP32–NF27 Page 3361
Pharmacopeial Forum: Volume No. 34(5) Page 1180
Chromatographic columns text is not derived from, and not part of, USP 32 or NF 27.