Pentetic Acid
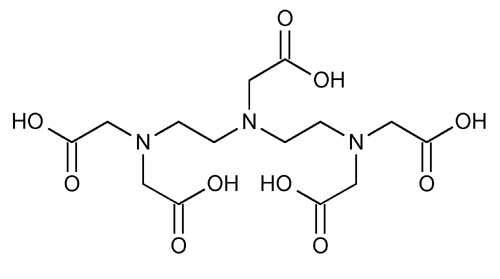
Glycine, N,N-bis[2-[bis(carboxymethyl)amino]ethyl].
Diethylenetriaminepentaacetic acid
» Pentetic Acid contains not less than 98.0 percent and not more than 100.5 percent of C14H23N3O10.
Packaging and storage—
Preserve in well-closed containers.
Identification, Infrared Absorption  197K
197K .
.
Residue on ignition 281:
not more than 0.2%.
Heavy metals, Method II 231:
0.005%.
Limit of nitrilotriacetic acid—
Cupric acetate solution—
Dissolve 20 g of cupric acetate in a mixture of 800 mL of water and 10 mL of glacial acetic acid. Adjust with 1 N sodium hydroxide to a pH of 4.2, dilute with water to obtain 1000 mL of solution, and filter.
Mobile phase—
Prepare a mixture of 1600 mL of water, 40 mL of glacial acetic acid, 30.4 mL of 0.5 M dodecyltriethylammonium phosphate, and 20 mL of Cupric acetate solution. Adjust with 1 N sodium hydroxide to a pH of 4.0, dilute with water to obtain 2000 mL of solution, filter through a filter having a 0.5-µm or finer porosity, and degas. Make adjustments if necessary (see System Suitability under Chromatography 621).
Stock standard solution—
Transfer about 50 mg of nitrilotriacetic acid, accurately weighed, to a 100-mL volumetric flask, dilute with Cupric acetate solution to volume, and mix.
Standard solution—
Transfer 1.0 mL of the Stock standard solution to a 25-mL volumetric flask, dilute with Cupric acetate solution to volume, and mix. This solution contains about 0.02 mg of nitrilotriacetic acid per mL.
Test solution—
Transfer about 2 g of Pentetic Acid, accurately weighed, to a 100-mL volumetric flask. Add about 70 mL of Cupric acetate solution, and swirl to dissolve. Sonicate, if necessary, to dissolve. Dilute with Cupric acetate solution to volume, and mix.
Resolution solution—
Transfer 1.0 mL of the Stock standard solution to a 25-mL volumetric flask, dilute with Test solution to volume, and mix.
Chromatographic system
(see Chromatography 621)—The liquid chromatograph is equipped with a 290-nm detector and a 4.6-mm × 25-cm column that contains 5-µm packing L1 that has been highly deactivated (carbon loading of about 30%). The flow rate is about 1 mL per minute. Equilibrate the column by passing, in sequence, water, methanol, and water for about 15 minutes each, and then Mobile phase for about 45 minutes. Chromatograph the Resolution solution, and record the peak responses as directed for Procedure: the resolution, R, between pentetic acid and nitrilotriacetic acid is not less than 2.0, and the relative retention times are about 0.6 for pentetic acid and 1.0 for nitrilotriacetic acid. Chromatograph the Standard solution, and record the peak responses as directed for Procedure: the relative standard deviation for replicate injections is not more than 5.0%.
Procedure—
Separately inject equal volumes (about 20 µL) of the Standard solution and the Test solution into the chromatograph, and measure the responses for the major peaks. Calculate the percentage of nitrilotriacetic acid in the portion of Pentetic Acid taken by the formula:
10,000(C / W)(rU / rS)
of which C is the concentration, in mg per mL, of nitrilotriacetic acid in the Standard solution, W is the weight, in mg, of Pentetic Acid taken to prepare the Test solution, and rU and rS are the nitrilotriacetic acid peak responses obtained from the Test solution and the Standard solution, respectively. The limit is 0.1%.
Iron—
Using 1.5 g of specimen, proceed as directed in the test for Iron under Edetic Acid. The color of the test solution is not deeper than that of the solution containing the standard iron solution (0.01%).
Assay—
Transfer about 200 mg of Pentetic Acid, accurately weighed, to a 125-mL conical flask, add 50 mL of water and 1.5 mL of 1 N sodium hydroxide, and swirl to dissolve the specimen. Add 10 mL of 0.1 N ammonium thiocyanate, and mix. Add about 40 mL of methyl ethyl ketone, mix, and allow the layers to separate. Titrate with 0.05 N ferric ammonium sulfate VS, stirring continuously. As the titration proceeds, the aqueous phase turns from colorless to yellow, and the organic phase remains colorless. As the endpoint is approached, stop the titration, mix, and allow the layers to separate. Add 0.1-mL increments of 0.05 N ferric ammonium sulfate VS, mixing and allowing the layers to separate after each addition, until the organic layer turns from colorless to pink. Each mL of 0.05 N ferric ammonium sulfate consumed is equivalent to 19.668 mg of C14H23N3O10.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
Chromatographic Column—
| Topic/Question | Contact | Expert Committee |
| Monograph | Elena Gonikberg, Ph.D.
Senior Scientist 1-301-816-8251 |
(MDGRE05) Monograph Development-Gastrointestinal Renal and Endocrine |
| Reference Standards | Lili Wang, Technical Services Scientist 1-301-816-8129 RSTech@usp.org |
USP32–NF27 Page 3248
Chromatographic columns text is not derived from, and not part of, USP 32 or NF 27.