Galantamine Tablets
» Galantamine Tablets contain an amount of Galantamine Hydrobromide equivalent to not less than 90.0 percent and not more than 110.0 percent of the labeled amount of galantamine (C17H21NO3).
Packaging and storage—
Preserve in well-closed containers, and store at controlled room temperature.
USP Reference standards  11
11 —
—
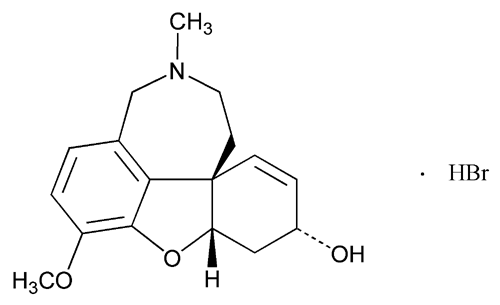
USP Galantamine Hydrobromide RS .
.
USP Galantamine Hydrobromide Related Compounds Mixture RS.
USP Galantamine Hydrobromide RS
 .
.
USP Galantamine Hydrobromide Related Compounds Mixture RS.
Identification—
A: Ultraviolet Absorption 197U
—The spectrum of the Test solution corresponds to that of the Standard solution, as obtained in the test for Uniformity of dosage units.
B:
The retention time of the major peak in the chromatogram of the Assay preparation corresponds to that in the chromatogram of the Standard preparation, as obtained in the Assay.
Dissolution 711—
Medium:
water; 500 mL.
Apparatus 2:
50 rpm.
Time:
20 minutes.
Standard solution—
Dissolve an accurately weighed quantity of USP Galantamine Hydrobromide RS in Medium, and dilute quantitatively, and stepwise if necessary, to obtain a solution having a known concentration of about 0.008 mg per mL of galantamine for Tablets labeled to contain 4 mg; about 0.016 mg per mL of galantamine for Tablets labeled to contain 8 mg; and about 0.024 mg per mL of galantamine for Tablets labeled to contain 12 mg. [note—The concentration of galantamine (free base), in mg per mL, can be calculated using the molecular weights of galantamine (287.35) and galantamine hydrobromide (368.27)]
Test solution—
Use portions of the solution under test passed through a suitable 0.2-µm filter.
Procedure—
Determine the amount of galantamine dissolved by employing UV absorption at the wavelength of maximum absorbance at about 288 nm on the Test solution in comparison with the Standard solution, using a 5-cm cell for Tablets labeled to contain 4 mg or 8 mg, or using a 1-cm cell for Tablets labeled to contain 12 mg. Calculate the percentage of galantamine (C17H21NO3) dissolved, by the formula:
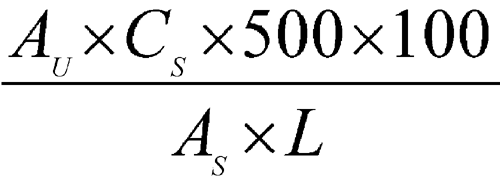 in which AU and AS are the absorbances obtained from the Test solution and the Standard solution, respecetively; CS is the concentration of galantamine, in mg per mL, in the Standard solution; 500 is the volume, in mL, of Medium; 100 is the conversion factor to percentage; and L is the Tablet label claim in mg.
in which AU and AS are the absorbances obtained from the Test solution and the Standard solution, respecetively; CS is the concentration of galantamine, in mg per mL, in the Standard solution; 500 is the volume, in mL, of Medium; 100 is the conversion factor to percentage; and L is the Tablet label claim in mg.
Tolerances—
Not less than 80% (Q) of the labeled amount of galantamine is dissolved in 20 minutes.
Uniformity of dosage units 905—
meets the requirements for coated tablets.
Standard solution—
Dissolve an accurately weighed quantity of USP Galantamine Hydrobromide RS in a suitable volumetric flask, and dilute quantitatively with 0.1 N hydrochloric acid to obtain a solution having a known concentration of about 0.04 mg of galantamine per mL. [note—The concentration of galantamine (free base), in mg per mL, can be calculated using the molecular weights of galantamine (287.35) and galantamine hydrobromide (368.27).]
Test solution—
Add one Tablet to each appropriately sized volumetric flask to obtain a final galantamine concentration of 0.04 mg per mL, add an appropriate amount of 0.1 N hydrochloric acid equivalent to 75% of the total volume of the volumetric flask, and mechanically shake for about 45 minutes. Dilute with 0.1 N hydrochloric acid to volume, and mix. Pass a portion of this solution through a filter having a 0.2-µm or finer porosity, and use the filtrate.
Procedure—
Determine the amount of galantamine (C17H21NO3) dissolved by employing UV absorption at the wavelength of maximum absorbance at about 289 nm on filtered portions of the Test solution in comparison with a Standard solution. Calculate the quantity of galantamine (C17H21NO3) dissolved, in percent of the label claim, by the formula:
(CS / CU)(AU / AS)100
in which CS is the concentration, in mg per mL, of galantamine in the Standard solution; CU is the concentration, in mg per mL, of galantamine in the Test solution based on the label claim; and AU and AS are the absorbances at the monitoring wavelength, obtained from the Test solution and the Standard solution, respectively.
Related compounds—
Buffer solution, Solution A, Solution B, Mobile phase, and Diluent—
Prepare as directed in the Assay.
Resolution solution—
Prepare a solution of USP Galantamine Hydrobromide Related Compounds Mixture RS in Diluent having a concentration of 0.6 mg per mL.
Standard solution—
Use the Standard preparation, prepared as directed in the Assay.
Test solution—
Use the Assay preparation.
Chromatographic system—
Prepare as directed in the Assay. Chromatograph about 20 µL of the Resolution solution, and record the responses as directed for Procedure. Identify the impurities using the approximate relative retention times given in Table 1: the resolution, R, between 6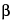 -hexahydrogalantamine and 6-octahydrogalantamine is not less than 1.5. Chromatograph the Standard solution, and record the responses as directed for Procedure: the relative standard deviation for replicate injections is not more than 2.0% for the galantamine peak.
-hexahydrogalantamine and 6-octahydrogalantamine is not less than 1.5. Chromatograph the Standard solution, and record the responses as directed for Procedure: the relative standard deviation for replicate injections is not more than 2.0% for the galantamine peak.
Procedure—
Separately inject equal volumes (about 20 µL) of the Standard solution and the Test solution into the chromatograph, record the chromatograms, and measure the peak responses. [note—Ignore the peak due to bromide near the void volume.] Calculate the percentage of each of the galantamine related compounds in the portion of Tablets taken by the formula:
100(Cs / CU)(rU / rS)(1/F)
in which CS and CU are the concentrations, in mg per mL, of galantamine in the Standard solution and Test solution, respectively; rU is the peak area of each impurity obtained from the Test solution; rS is the peak area of galantamine obtained from the Standard solution; and F is the Relative Response Factor (see Table 1 for values) for each of the impurities relative to galantamine.
Table 1
| Related Compound | Relative Retention Time (RRT) |
Relative Response Factor (F) |
Limit % |
| 6 |
0.73 | 1.1 | 0.30 |
| 6 |
0.86 | — | NA |
| Galantamine hydrobromide | 1.00 | 1.0 | NA |
| 6 |
1.15 | — | NA |
| Tetrahydrogalantamine†4 | 2.09 | — | NA |
| Individual unspecified degradation impurity | — | 1.0 | 0.20% |
| Total impurities | — | — | 1.0% |
|
[note—The impurities marked with “†” are not quantified and are intended for system suitability evaluation only.]
1
[4aS-(4
2
[4aS-(4a
3
[4aS-(4a
4
[4aS-(4aR*,8aR*)]-9,10,11,12-Tetrahydro-3-methoxy-11-methyl-4aH-benzofuro[3a,3,2-ef][2]benzazepine.
|
|||
Assay—
Buffer solution–
Dissolve 5.34 g of dibasic sodium phosphate dihydrate in 1 L of water. Adjust with phosphoric acid to a pH of 6.5, and mix.
Solution A—
Add 950 mL of Buffer solution to 50 mL of methanol, and mix.
Solution B—
Prepare a mixture of acetonitrile and methanol (95:5).
Mobile phase—
Use variable mixtures of Solution A and Solution B as directed for Chromatographic system. Make adjustments if necessary (see System Suitability under Chromatography 621).
Diluent—
Dissolve about 35.4 g of edetate disodium in 950 mL of water. Add 50 mL of methanol, and mix well.
Standard preparation—
Dissolve an accurately weighed quantity of USP Galantamine Hydrobromide RS in Diluent, and dilute quantitatively, and stepwise if necessary, with Diluent to obtain a solution having a known concentration of about 0.48 mg per mL of galantamine. [note—The concentration of galantamine (free base), in mg per mL, can be calculated using the molecular weights of galantamine (287.35) and galantamine hydrobromide ( 368.27)]
Assay preparation—
Weigh not fewer than 10 Tablets. Transfer an accurately weighed portion to an appropriately-sized volumetric flask, and dilute quantitatively, and stepwise if necessary, with Diluent to obtain a solution having a galantamine concentration of about 0.48 mg per mL, based on the label claim. Pass a portion of this solution through a PTFE filter having a 0.45-µm or finer porosity, and use the filtrate.
Chromatographic system (see Chromatography 621)—
The liquid chromatograph is equipped with a 230-nm detector and a 4.6-mm × 10-cm column that contains 3-µm L1 packing. The flow rate is about 1.5 mL per minute. The column temperature is maintained at 35 . The chromatograph is programmed as follows.
. The chromatograph is programmed as follows.
Chromatograph the Standard preparation, and record the peak responses as directed for Procedure: the relative standard deviation for replicate injections is not more than 2.0%.
| Time (minutes) |
Solution A
(%) |
Solution B
(%) |
|
| 0.0–40.0 | 100®75 | 0®25 | linear gradient |
| 40.0–45.0 | 75®60 | 25®40 | linear gradient |
| 45.0–46.0 | 60®40 | 40®60 | linear gradient |
| 46.0–55.0 | 40 | 60 | isocratic |
| 55.0–56.0 | 40®100 | 60®0 | linear gradient |
| 56.0–61.0 | 100 | 0 | re-equilbration |
Procedure—
Separately inject equal volumes (about 20 µL) of the Standard preparation and the Assay preparation into the chromatograph, and measure the responses for the galantamine hydrobromide peak. Calculate the quantity, in percentage of label claim, of galantamine (C17H21NO3) in the portion of Tablets taken by the formula:
100(CS / CU)(rU / rS)
in which CS and CU are the concentrations of galantamine, in mg per mL, in the Standard preparation and the Assay preparation, respectively; and rU and rS are the peak responses obtained from the Assay preparation and the Standard preparation, respectively.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
Chromatographic Column—
| Topic/Question | Contact | Expert Committee |
| Monograph | Ravi Ravichandran, Ph.D.
Senior Scientist 1-301-816-8330 |
(MDPP05) Monograph Development-Psychiatrics and Psychoactives |
| Reference Standards | Lili Wang, Technical Services Scientist 1-301-816-8129 RSTech@usp.org |
|
| Margareth R.C. Marques, Ph.D.
Senior Scientist 1-301-816-8106 |
(BPC05) Biopharmaceutics05 |
USP32–NF27 Page 2478
Pharmacopeial Forum: Volume No. 34(6) Page 1452
Chromatographic columns text is not derived from, and not part of, USP 32 or NF 27.