Add the following:
» Cefdinir for Oral Suspension contains not less than 90.0 percent and not more than 110.0 percent of the labeled amount of C14H13N5O5S2. It may contain one or more suitable buffers, flavors, preservatives, stabilizing agents, sweeteners, and suspending agents.
Packaging and storage—
Preserve in tight, light-resistant containers, and store at controlled room temperature.
Labeling—
The label specifies the directions for the constitution of the powder and states the equivalent amount of C14H13N5O5S2 in a given volume of Oral Suspension after constitution.
USP Reference standards  11
11 —
—
USP Cefdinir RS.
USP Cefdinir Related Compound A RS.
USP Cefdinir Related Compound B RS.
USP Cefdinir RS.
USP Cefdinir Related Compound A RS.
USP Cefdinir Related Compound B RS.
Identification—
A: Thin-Layer Chromatographic Identification Test 201—
0.1 M Phosphate buffer solution—
Prepare as directed in the Assay.
Standard solution—
Transfer an appropriate amount of USP Cefdinir RS into a suitable volumetric flask. Dissolve in and dilute with methanol and 0.1 M Phosphate buffer solution (75:25) to obtain a solution containing a known amount of about 0.6 mg/mL of USP Cefdinir RS.
Test solution—
Mix a portion of constituted Oral Suspension equivalent to about 125 mg of Cefdinir in a 100-mL volumetric flask with 50 mL of 0.1 M Phosphate buffer solution, and dilute with methanol to volume. Pass a portion through a suitable 0.45-µm filter. Transfer 5.0 mL of the filtrate to a 10-mL volumetric flask, and dilute with methanol to volume.
Procedure—
Separately apply 10 µL of the Test solution and the Standard solution to a suitable thin-layer chromatographic plate (see Chromatography 621) coated with a 0.25-mm layer of chromatographic silica gel, preconditioned with n-hexane–tetradecane solution (95:5), and allow the spots to dry. Place the plate in a chamber equilibrated with a solvent mixture consisting of methanol and water (80:20), and develop the chromatogram in the same solvent system until the solvent front has moved about 15 cm. Remove the plate from the developing chamber, allow the solvent to evaporate, and visually locate the spots under a short-wavelength UV light: the RF value of the principal spot obtained from the Test solution corresponds to that obtained from the Standard solution.
B:
The retention time of the main peak in the chromatogram of the Assay preparation corresponds to that in the chromatogram of the Standard preparation, as obtained in the Assay.
Dissolution 711—
Medium:
0.05 M phosphate buffer, pH 6.8; 900 mL.
Apparatus 2:
50 rpm.
Time:
30 minutes.
Standard solution—
Dissolve an accurately weighed quantity of USP Cefdinir RS, and dilute with Medium, quantitatively, and stepwise, if necessary, to obtain a solution having a concentration of about 0.14 mg per mL.
Test solution—
Transfer 5 mL, by weight, of the reconstituted Oral Suspension into the vessel. After the appropriate time withdraw a portion of the solution under test and pass through a suitable 0.45-µm filter. Dilute with Medium a portion of each filtered sample as necessary to obtain a solution having a concentration of about 0.14 mg per mL of Cefdinir.
Procedure—
Determine the amount of C14H13N5O5S2 dissolved by employing UV absorption at the wavelength of maximum absorbance at about 290 nm on portions of the Test solution in comparison with the Standard solution, using Medium as blank. Calculate the percentage of cefdinir dissolved by the formula:
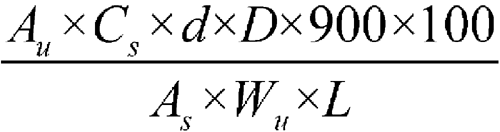 in which AU and AS are the absorbances obtained from the Test solution and Standard solution, respectively; CS is the concentration, in mg per mL, of cefdinir in the Standard solution; d is the density, in mg per mL, of the Oral Suspension obtained by dividing the weight of Oral Suspension taken by 5 mL; D is the dissolution factor used, if necessary, to prepare the Test solution; 900 is the volume, in mL, of Medium; 100 is the conversion factor to percentage; WU is the weight, in mg, of Oral Suspension taken; and L is the label claim, in mg.
in which AU and AS are the absorbances obtained from the Test solution and Standard solution, respectively; CS is the concentration, in mg per mL, of cefdinir in the Standard solution; d is the density, in mg per mL, of the Oral Suspension obtained by dividing the weight of Oral Suspension taken by 5 mL; D is the dissolution factor used, if necessary, to prepare the Test solution; 900 is the volume, in mL, of Medium; 100 is the conversion factor to percentage; WU is the weight, in mg, of Oral Suspension taken; and L is the label claim, in mg.
Tolerances—
Not less than 80% (Q) of the labeled amount of C14H13N5O5S2 is dissolved in 30 minutes.
Uniformity of dosage units 905—For Oral Suspension packaged in single-unit containers:
meets the requirements.
Deliverable volume 698—For Oral Suspension packaged in multiple-unit containers:
meets the requirements.
pH 791:
between 3.5 and 4.5.
Loss on drying 731—
Dry about 1 g of powder over phosphorous pentoxide in a vacuum oven not exceeding 5 mm of mercury at 70 for 4 to 4.5 hours: it loses not more than 1.0%.
for 4 to 4.5 hours: it loses not more than 1.0%.
Related compounds—
0.1 M Phosphate buffer solution—
solution A—
Dissolve in and dilute with water 14.2 g of anhydrous dibasic sodium phosphate to 1000.0 mL.
solution B—
Dissolve in and dilute with water 6.8 g of monobasic potassium phosphate to 500.0 mL.
solution C—
Mix 1000 mL of Solution A with 500 mL of Solution B. Verify a pH of 7.0 ± 0.1.
Dilute phosphoric acid solution—
Dilute phosphoric acid with water (1 in 10), and mix.
0.1 M Disodium ethylenediaminetetraacetate (EDTA)—
Transfer about 3.72 g of disodium ethylenediaminetetraacetate into a 100-mL volumetric flask. Dissolve in and dilute with water to volume, and mix.
0.1% Tetramethylammonium hydroxide solution—
Dilute 20 mL of tetramethylammonium hydroxide (10% in water) with water to make 2000 mL, and mix. Adjust with Dilute phosphoric acid solution to a pH of 5.5 ± 0.1.
Mobile phase A—
Transfer 0.4 mL of 0.1 M EDTA to 1000 mL of 0.1% Tetramethylammonium hydroxide solution, and mix.
Mobile phase B—
Mix 250 mL of 0.1% Tetramethylammonium hydroxide solution, 150 mL of acetonitrile, and 100 mL of methanol. Add 0.2 mL of 0.1 M EDTA, and mix.
Mobile phase—
Use variable mixtures of Mobile phase A and Mobile phase B as directed in the Chromatographic system.
Standard solution 1—
Dissolve an appropriate amount of USP Cefdinir RS in 0.1 M Phosphate buffer solution (Solution C) to obtain a solution having a known concentration of about 0.75 mg per mL of USP Cefdinir RS. Dilute with 0.1% Tetramethyl ammonium hydroxide solution an appropriate amount of this solution, stepwise, if necessary, to obtain a solution having a known concentration of about 15 µg per mL.
Standard solution 2—
Dissolve an appropriate quantity of USP Cefdinir Related Compound A RS in 0.1% Tetramethylammonium hydroxide solution to obtain a solution having a known concentration of about 0.04 mg per mL.
Standard solution 3—
Dissolve an appropriate quantity of USP Cefdinir Related Compound B RS in 0.1 M Phosphate buffer solution to obtain a solution having a known concentration of about 0.04 mg per mL.
Resolution solution—
Transfer about 37.5 mg of accurately weighed USP Cefdinir RS into a 25-mL volumetric flask. Add about 10 mL of 0.1 M Phosphate buffer solution. Add 5.0 mL of each of Standard solution 2 and Standard solution 3, and dilute with 0.1% Tetramethylammonium hydroxide solution to volume.
Test solution—
Transfer a volume of constituted Oral Suspension equivalent to about 150 mg of Cefdinir into a 100-mL volumetric flask. Dissolve in 30 mL of 0.1 M Phosphate buffer solution (Solution C), and dilute with 0.1% Tetramethylammonium hydroxide solution to volume.
Chromatographic system (see Chromatography 621)—
The liquid chromatograph is equipped with a 254-nm detector and a 4.6-mm × 150-mm column that contains 5-µm packing L1. The Test solution is maintained at a temperature of 4 ± 3, and the column temperature is maintained at 40 ± 0.5. The flow rate is about 1 mL per minute. The chromatograph is programmed as follows for a run time of about 60 minutes.
| Time (minutes) |
Mobile phase A (%) | Mobile phase B (%) | |
| 0–2 | 95 | 5 | isocratic |
| 2–22 | 95®75 | 5®25 | linear gradient |
| 22–32 | 75®50 | 25®50 | linear gradient |
| 32–37 | 50 | 50 | isocratic |
| 37–38 | 50®95 | 50®5 | linear gradient |
| 38–58 | 95 | 5 | isocratic |
Procedure—
Separately inject equal volumes (about 10 µL) of the Resolution solution, Standard solution 1, and the Test solution into the chromatograph, record the chromatograms, and measure the peak responses: the resolution between the cefdinir peak and the third peak of cefdinir related compound A is greater than 1.5; the tailing factor of cefdinir related compound B is not more than 1.5; and the relative standard deviation for the cefdinir peak response in replicate injections of Standard solution 1 is not more than 2.0%. The approximate relative retention times and the relative response factors for each of the related compounds compared to cefdinir are given in Table 1. Calculate the percentage of individual specified and unspecified impurities in the portion of Oral Suspension taken by the formula:
(100 / F)(CS / CU)(rU / rS)
in which F is the relative response factor for each impurity, as mentioned in Table 1; CS is the concentration, in mg per mL, of USP Cefdinir RS in Standard solution 1; CU is the concentration, in mg per mL, of Cefdinir in the Test solution; rU is the peak response of each impurity obtained from the Test solution; and rS is the peak response of cefdinir obtained from Standard solution 1. The specified and unspecified impurities meet the limits specified in Table 1.
Table 1
| Related Compound | Approximate Relative Retention Time |
Relative Response Factor |
Limit of Quantitation (% of Cefdinir) |
Limit (%) |
| Impurity VIII | 0.10 | 1.1 | 0.1 | NMT 0.5 |
| Impurity IV | 0.13 | 1.1 | 0.1 | NMT 0.6 |
| Impurity XIV | 0.36 | 1.0 | 0.05 | NMT 0.2 |
| Impurity V | 0.46 | 1.5 | 0.05 | NMT 0.3 |
| Impurity B3 | 0.77 | 1.0 | 0.05 | NMT 0.2 |
| Impurity XI | 0.75 | 1.0 | 0.05 | NMT 0.7 |
| Cefdinir related compound A1
(Isomer a) |
0.85 | 1.5 | 0.1 | NMT 3.3 |
| Cefdinir related compound A1
(Isomer b) |
0.94 | 1.5 | 0.1 | — |
| Cefdinir related compound A1
(Isomer c) |
1.11 | 1.5 | 0.1 | — |
| Cefdinir related compound A1
(Isomer d) |
1.14 | 1.5 | 0.1 | — |
| Impurity VI | 1.18 | 1.1 | 0.05 | NMT 0.2 |
| Impurity I | 1.23 | 1.2 | 0.05 | NMT 0.8 |
| Cefdinir related compound B | 1.28 | 1.1 | 0.05 | NMT 0.2 |
| Impurity XIII | 1.37 | 1.4 | 0.05 | NMT 0.5 |
| Impurity E3 | 1.44 | 1.0 | 0.05 | NMT 0.2 |
| Impurity XV | 1.49 | 1.0 | 0.05 | NMT 0.2 |
| Impurity VII | 1.51 | 1.1 | 0.05 | NMT 1.2 |
| Impurity IIIa2 | 1.62 | 1.3 | 0.05 | NMT 1.1 |
| Impurity IIIb2 | 1.64 | 1.3 | 0.05 | — |
| Impurity D3 | 1.82 | 1.0 | 0.05 | NMT 0.2 |
| Individual unidentified impurities |
N/A | 1.0 | 0.05 | NMT 0.2 |
| Total unidentified impurities4 | N/A | N/A | N/A | NMT 0.9 |
| Total impurities | N/A | N/A | N/A | NMT 6.2 |
|
1
Cefdinir related compound A is a mixture of 4 isomers designated as lactam ring cleavage lactones a, b, c, and d. The sum of all values is reported, and the total limit for all 4 isomers combined is 3.3%.
2
Impurity III is a mixture of 2 isomers designated as Impurity IIIa and Impurity IIIb. The sum of both values is reported, and the total limit for both isomers combined is 1.5%.
3
Impurity B, Impurity D, and Impurity E are unidentified impurities.
4
The total unidentified impurities limit includes the % total of unidentified impurities B, D, and E and any other unidentified impurities detected.
|
||||
Assay—
0.1 M Phosphate buffer solution—
Dissolve 10.65 g of anhydrous dibasic sodium phosphate and 3.40 g of monobasic potassium phosphate in 750 mL of water. Adjust with phosphoric acid or sodium hydroxide to a pH of 7.0 ± 0.05, and dilute with water to 1000 mL.
Citric acid buffer solution—
Dissolve 7.0 g of citric acid monohydrate in 1000 mL of water, and adjust with phosphoric acid to a pH of 2.0 ± 0.05.
Mobile phase—
Mix 1000 mL of Citric acid buffer solution with 111 mL of methanol and 28 mL of tetrahydrofuran. Make adjustments if necessary (see System Suitability under Chromatography 621).
Standard preparation—
Accurately weigh a known amount of USP Cefdinir RS into a suitable volumetric flask. Dissolve in 0.1 M Phosphate buffer solution to obtain a solution having a known concentration of about 0.05 mg per mL of cefdinir.
Assay preparation—
Quantitatively transfer the contents of each bottle of constituted Oral Suspension into a suitable flask, and dilute with 0.1 M Phosphate buffer solution to obtain a solution having a concentration of about 0.05 mg per mL of cefdinir.
Resolution solution—
Accurately weigh known quantities of USP Cefdinir RS and m-hydroxybenzoic acid into a suitable volumetric flask. Dissolve in 0.1 M Phosphate buffer solution to obtain a solution having a known concentration of about 0.05 mg per mL of cefdinir and about 0.175 mg per mL of m-hydroxybenzoic acid.
Chromatographic system
(see Chromatography 621)—The liquid chromatograph is equipped with a 254-nm detector and a 3.9-mm × 150-mm column that contains 4-µm packing L1. The flow rate is maintained at about 1.4 mL per minute. Chromatograph the Resolution solution, and record the peak response as directed in the Procedure. The resolution between cefdinir and m-hydroxybenzoic acid is greater than 3.0; the tailing factor of the cefdinir peak is not more than 2.0; and the relative standard deviation for replicate injections of the Standard preparation is not more than 1.0%.
Procedure—
Separately inject equal volumes (about 15 µL) of the Standard preparation and the Assay preparation into the chromatograph, record the chromatograms, and measure the responses for the major peaks. Calculate the percentage of cefdinir (C14H13N5O5S2) in the portion of Oral Suspension taken by the formula:
 USP32
USP32
100(CS / CU)(rU / rS)
in which CS is the concentration, in mg per mL, of cefdinir in the Standard solution; CU is the concentration of cefdinir in the Assay preparation; and rU and rS are the peak responses of cefdinir in the Assay preparation and the Standard preparation, respectively.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
Chromatographic Column—
| Topic/Question | Contact | Expert Committee |
| Monograph | Ahalya Wise, M.S.
Scientist 1-301-816-8161 |
(MDANT05) Monograph Development-Antibiotics |
| Reference Standards | Lili Wang, Technical Services Scientist 1-301-816-8129 RSTech@usp.org |
|
| Margareth R.C. Marques, Ph.D.
Senior Scientist 1-301-816-8106 |
(BPC05) Biopharmaceutics05 |
USP32–NF27 Page 1830
Pharmacopeial Forum: Volume No. 34(1) Page 81
Chromatographic columns text is not derived from, and not part of, USP 32 or NF 27.