INTRODUCTION
The objective of this chapter is to describe conditions and practices to prevent harm, including death, to patients that could result from (1) microbial contamination (nonsterility), (2) excessive bacterial endotoxins, (3) variability in the intended strength of correct ingredients that exceeds either monograph limits for official articles (see “official” and “article” in the General Notices and Requirements) or 10% for nonofficial articles, (4) unintended chemical and physical contaminants, and (5) ingredients of inappropriate quality in compounded sterile preparations (CSPs). Contaminated CSPs are potentially most hazardous to patients when administered into body cavities, central nervous and vascular systems, eyes, and joints, and when used as baths for live organs and tissues. When CSPs contain excessive bacterial endotoxins (see Bacterial Endotoxins Test  85
85 ), they are potentially most hazardous to patients when administered into the central nervous system.
), they are potentially most hazardous to patients when administered into the central nervous system.
Despite the extensive attention in this chapter to the provision, maintenance, and evaluation of air quality, the avoidance of direct or physical contact contamination is paramount. It is generally acknowledged that direct or physical contact of critical sites of CSPs with contaminants, especially microbial sources, poses the greatest probability of risk to patients. Therefore, compounding personnel must be meticulously conscientious in precluding contact contamination of CSPs both within and outside ISO Class 5 (see Table 1) areas.
To achieve the above five conditions and practices, this chapter provides minimum practice and quality standards for CSPs of drugs and nutrients based on current scientific information and best sterile compounding practices. The use of technologies, techniques, materials, and procedures other than those described in this chapter is not prohibited so long as they have been proven to be equivalent or superior with statistical significance to those described herein. The standards in this chapter do not pertain to the clinical administration of CSPs to patients via application, implantation, infusion, inhalation, injection, insertion, instillation, and irrigation, which are the routes of administration. Four specific categories of CSPs are described in this chapter: low-risk level, medium-risk level, and high-risk level, and immediate use. Sterile compounding differs from nonsterile compounding (see Pharmaceutical Compounding—Nonsterile Preparations 795 and Good Compounding Practices 1075) primarily by requiring the maintenance of sterility when compounding exclusively with sterile ingredients and components (i.e., with immediate-use CSPs, low-risk level CSPs, and medium-risk level CSPs) and the achievement of sterility when compounding with nonsterile ingredients and components (i.e., with high-risk level CSPs). Some differences between standards for sterile compounding in this chapter and those for nonsterile compounding in Pharmaceutical Compounding—Nonsterile Preparations 795 include, but are not limited to, ISO-classified air environments (see Table 1); personnel garbing and gloving; personnel training and testing in principles and practices of aseptic manipulations and sterilization; environmental quality specifications and monitoring; and disinfection of gloves and surfaces of ISO Class 5 (see Table 1) sources.
Table 1. ISO Classification of Particulate Matter in Room Air (limits are in particles of 0.5 µm and larger per cubic meter [current ISO] and cubic feet [former Federal Standard No. 209E, FS 209E])*
| Class Name | Particle Count | ||
| ISO Class | U.S. FS 209E | ISO, m3 | FS 209E, ft3 |
| 3 | Class 1 | 35.2 | 1 |
| 4 | Class 10 | 352 | 10 |
| 5 | Class 100 | 3,520 | 100 |
| 6 | Class 1,000 | 35,200 | 1,000 |
| 7 | Class 10,000 | 352,000 | 10,000 |
| 8 | Class 100,000 | 3,520,000 | 100,000 |
|
*
Adapted from former Federal Standard No. 209E, General Services Administration, Washington, DC, 20407 (September 11, 1992) and ISO 14644-1:1999, Cleanrooms and associated controlled environments—Part 1: Classification of air cleanliness. For example, 3,520 particles of 0.5 µm per m3 or larger (ISO Class 5) is equivalent to 100 particles per ft3 (Class 100) (1 m3 = 35.2 ft3).
|
|||
The standards in this chapter are intended to apply to all persons who prepare CSPs and all places where CSPs are prepared (e.g., hospitals and other healthcare institutions, patient treatment clinics, pharmacies, physicians' practice facilities, and other locations and facilities in which CSPs are prepared, stored, and transported). Persons who perform sterile compounding include pharmacists, nurses, pharmacy technicians, and physicians. These terms recognize that most sterile compounding is performed by or under the supervision of pharmacists in pharmacies and also that this chapter applies to all healthcare personnel who prepare, store, and transport CSPs. For the purposes of this chapter, CSPs include any of the following:
- Compounded biologics, diagnostics, drugs, nutrients, and radiopharmaceuticals, including but not limited to the following dosage forms that must be sterile when they are administered to patients: aqueous bronchial and nasal inhalations, baths and soaks for live organs and tissues, injections (e.g., colloidal dispersions, emulsions, solutions, suspensions), irrigations for wounds and body cavities, ophthalmic drops and ointments, and tissue implants.
-
Manufactured sterile products that are either prepared strictly according to the instructions appearing in manufacturers' approved labeling (product package inserts) or prepared differently than published in such labeling. [note—The FDA states that “Compounding does not include mixing, reconstituting, or similar acts that are performed in accordance with the directions contained in approved labeling provided by the product's manufacturer and other manufacturer directions consistent with that labeling” [21 USC 321 (k) and (m)]. However, the FDA-approved labeling (product package insert) rarely describes environmental quality (e.g., ISO Class air designation, exposure durations to non-ISO classified air, personnel garbing and gloving, and other aseptic precautions by which sterile products are to be prepared for administration). Beyond-use exposure and storage dates or times (see General Notices and Requirements and Pharmaceutical Compounding—Nonsterile Preparations 795) for sterile products that have been either opened or prepared for administration are not specified in all package inserts for all sterile products. Furthermore, when such durations are specified, they may refer to chemical stability and not necessarily to microbiological purity or safety.]
ORGANIZATION OF THIS CHAPTER
The sections in this chapter are organized to facilitate the practitioner's understanding of the fundamental accuracy and quality practices for preparing CSPs. They provide a foundation for the development and implementation of essential procedures for the safe preparation of low-risk, medium-risk, and high-risk level CSPs and immediate-use CSPs, which are classified according to the potential for microbial, chemical, and physical contamination. The chapter is divided into the following main sections:
- Definitions
- Responsibility of Compounding Personnel
- CSP Microbial Contamination Risk Levels
- Personnel Training and Evaluation in Aseptic Manipulation Skills
- Immediate-Use CSPs
- Single-Dose and Multiple-Dose Containers
- Hazardous Drugs as CSPs
- Radiopharmaceuticals as CSPs
- Allergen Extracts as CSPs
- Verification of Compounding Accuracy and Sterility
- Environmental Quality and Control
- Suggested Standard Operating Procedures (SOPs)
- Elements of Quality Control
- Verification of Automated Compounding Devices (ACDs) for Parenteral Nutrition Compounding
- Finished Preparation Release Checks and Tests
- Storage and Beyond-Use Dating
- Maintaining Sterility, Purity, and Stability of Dispensed and Distributed CSPs
- Patient or Caregiver Training
- Patient Monitoring and Adverse Events Reporting
- Quality Assurance (QA) Program
- Abbreviations and Acronyms
- Appendices I–V
The requirements and recommendations in this chapter are summarized in Appendix I. A list of abbreviations and acronyms is included at the end of the main text, before the Appendices.
All personnel who prepare CSPs shall be responsible for understanding these fundamental practices and precautions, for developing and implementing appropriate procedures, and for continually evaluating these procedures and the quality of final CSPs to prevent harm.
DEFINITIONS
Ante-Area—
An ISO Class 8 (see Table 1) or better area where personnel hand hygiene and garbing procedures, staging of components, order entry, CSP labeling, and other high-particulate-generating activities are performed. It is also a transition area that (1) provides assurance that pressure relationships are constantly maintained so that air flows from clean to dirty areas and (2) reduces the need for the heating, ventilating, and air-conditioning (HVAC) control system to respond to large disturbances.1
Aseptic Processing
(see Microbiological Evaluation of Clean Rooms and Other Controlled Environments 1116)—A mode of processing pharmaceutical and medical products that involves the separate sterilization of the product and of the package (containers–closures or packaging material for medical devices) and the transfer of the product into the container and its closure under at least ISO Class 5 (see Table 1) conditions.
Beyond-Use Date (BUD)
(see General Notices and Requirements and Pharmaceutical Compounding—Nonsterile Preparations 795)—For the purpose of this chapter, the date or time after which a CSP shall not be stored or transported. The date is determined from the date or time the preparation is compounded.
Biological Safety Cabinet (BSC)—
A ventilated cabinet for CSPs, personnel, product, and environmental protection having an open front with inward airflow for personnel protection, downward high-efficiency particulate air (HEPA)-filtered laminar airflow for product protection, and HEPA-filtered exhausted air for environmental protection.
Buffer Area—
An area where the primary engineering control (PEC) is physically located. Activities that occur in this area include the preparation and staging of components and supplies used when compounding CSPs.
Clean Room
(see Microbiological Evaluation of Clean Rooms and Other Controlled Environments 1116 and also the definition of Buffer Area)—A room in which the concentration of airborne particles is controlled to meet a specified airborne particulate cleanliness class. Microorganisms in the environment are monitored so that a microbial level for air, surface, and personnel gear are not exceeded for a specified cleanliness class.
Compounding Aseptic Containment Isolator (CACI)—
A compounding aseptic isolator (CAI) designed to provide worker protection from exposure to undesirable levels of airborne drug throughout the compounding and material transfer processes and to provide an aseptic environment for compounding sterile preparations. Air exchange with the surrounding environment should not occur unless the air is first passed through a microbial retentive filter (HEPA minimum) system capable of containing airborne concentrations of the physical size and state of the drug being compounded. Where volatile hazardous drugs are prepared, the exhaust air from the isolator should be appropriately removed by properly designed building ventilation.
Compounding Aseptic Isolator (CAI)—
A form of isolator specifically designed for compounding pharmaceutical ingredients or preparations. It is designed to maintain an aseptic compounding environment within the isolator throughout the compounding and material transfer processes. Air exchange into the isolator from the surrounding environment should not occur unless the air has first passed through a microbially retentive filter (HEPA minimum).2
Critical Area—
An ISO Class 5 (see Table 1) environment.
Critical Site—
A location that includes any component or fluid pathway surfaces (e.g., vial septa, injection ports, beakers) or openings (e.g., opened ampuls, needle hubs) exposed and at risk of direct contact with air (e.g., ambient room or HEPA filtered), moisture (e.g., oral and mucosal secretions), or touch contamination. Risk of microbial particulate contamination of the critical site increases with the size of the openings and exposure time.
Direct Compounding Area (DCA)—
A critical area within the ISO Class 5 (see Table 1) primary engineering control (PEC) where critical sites are exposed to unidirectional HEPA-filtered air, also known as first air.
Disinfectant—
An agent that frees from infection, usually a chemical agent but sometimes a physical one, and that destroys disease-causing pathogens or other harmful microorganisms but may not kill bacterial and fungal spores. It refers to substances applied to inanimate objects.
First Air—
The air exiting the HEPA filter in a unidirectional air stream that is essentially particle free.
Hazardous Drugs—
Drugs are classified as hazardous if studies in animals or humans indicate that exposures to them have a potential for causing cancer, development or reproductive toxicity, or harm to organs. (See current NIOSH publication.)
Labeling
[see General Notices and Requirements and 21 USC 321 (k) and (m)]—A term that designates all labels and other written, printed, or graphic matter on an immediate container of an article or preparation or on, or in, any package or wrapper in which it is enclosed, except any outer shipping container. The term “label” designates that part of the labeling on the immediate container.
Media-Fill Test
(see Microbiological Evaluation of Clean Rooms and Other Controlled Environments 1116)—A test used to qualify aseptic technique of compounding personnel or processes and to ensure that the processes used are able to produce sterile product without microbial contamination. During this test, a microbiological growth medium such as Soybean–Casein Digest Medium is substituted for the actual drug product to simulate admixture compounding.3 The issues to consider in the development of a media-fill test are media-fill procedures, media selection, fill volume, incubation, time and temperature, inspection of filled units, documentation, interpretation of results, and possible corrective actions required.
Multiple-Dose Container
(see General Notices and Requirements and Containers for Injections under Injections 1)—A multiple-unit container for articles or preparations intended for parenteral administration only and usually containing antimicrobial preservatives. The beyond-use date (BUD) for an opened or entered (e.g., needle-punctured) multiple-dose container with antimicrobial preservatives is 28 days (see Antimicrobial Effectiveness Testing 51), unless otherwise specified by the manufacturer.
Negative Pressure Room—
A room that is at a lower pressure than the adjacent spaces and, therefore, the net flow of air is into the room.1
Pharmacy Bulk Package
(see Containers for Injections under Injections 1)—A container of a sterile preparation for parenteral use that contains many single doses. The contents are intended for use in a pharmacy admixture program and are restricted to the preparation of admixtures for infusion or, through a sterile transfer device, for the filling of empty sterile syringes. The closure shall be penetrated only one time after constitution with a suitable sterile transfer device or dispensing set, which allows measured dispensing of the contents. The pharmacy bulk package is to be used only in a suitable work area such as a laminar flow hood (or an equivalent clean air compounding area).
Where a container is offered as a pharmacy bulk package, the label shall (a) state prominently “Pharmacy Bulk Package—Not for Direct Infusion,” (b) contain or refer to information on proper techniques to help ensure safe use of the product, and (c) bear a statement limiting the time frame in which the container may be used once it has been entered, provided it is held under the labeled storage conditions.
Primary Engineering Control (PEC)—
A device or room that provides an ISO Class 5 (see Table 1) environment for the exposure of critical sites when compounding CSPs. Such devices include, but may not be limited to, laminar airflow workbenches (LAFWs), biological safety cabinets (BSCs), compounding aseptic isolators (CAIs), and compounding aseptic containment isolators (CACIs).
Preparation—
A preparation, or a CSP, that is a sterile drug or nutrient compounded in a licensed pharmacy or other healthcare-related facility pursuant to the order of a licensed prescriber; the article may or may not contain sterile products.
Product—
A commercially manufactured sterile drug or nutrient that has been evaluated for safety and efficacy by the FDA. Products are accompanied by full prescribing information, which is commonly known as the FDA-approved manufacturer's labeling or product package insert.
Positive Pressure Room—
A room that is at a higher pressure than the adjacent spaces and, therefore, the net airflow is out of the room.1
Single-Dose Container
(see General Notices and Requirements and Containers for Injections under Injections 1)—A single-dose container is a single-unit container for articles (see General Notices and Requirements) or preparations intended for parenteral administration only. It is intended for a single use. A single-dose container is labeled as such. Examples of single-dose containers include prefilled syringes, cartridges, fusion-sealed containers, and closure-sealed containers when so labeled.
Segregated Compounding Area—
A designated space, either a demarcated area or room, that is restricted to preparing low-risk level CSPs with 12-hour or less BUD. Such area shall contain a device that provides unidirectional airflow of ISO Class 5 (see Table 1) air quality for preparation of CSPs and shall be void of activities and materials that are extraneous to sterile compounding.
Sterilizing Grade Membranes—
Membranes that are documented to retain 100% of a culture of 107 microorganisms of a strain of Brevundimonas (Pseudomonas) diminuta per square centimeter of membrane surface under a pressure of not less than 30 psi (2.0 bar). Such filter membranes are nominally at 0.22-µm or 0.2-µm nominal pore size, depending on the manufacturer's practice.
Sterilization by Filtration—
Passage of a fluid or solution through a sterilizing grade membrane to produce a sterile effluent.
Terminal Sterilization—
The application of a lethal process (e.g., steam under pressure or autoclaving) to sealed containers for the purpose of achieving a predetermined sterility assurance level of usually less than 10 6, or a probability of less than one in one million of a nonsterile unit.3
6, or a probability of less than one in one million of a nonsterile unit.3
Unidirectional Flow
(see footnote 3)—An airflow moving in a single direction in a robust and uniform manner and at sufficient speed to reproducibly sweep particles away from the critical processing or testing area.
RESPONSIBILITY OF COMPOUNDING PERSONNEL
Compounding personnel are responsible for ensuring that CSPs are accurately identified, measured, diluted, and mixed and are correctly purified, sterilized, packaged, sealed, labeled, stored, dispensed, and distributed. These performance responsibilities include maintaining appropriate cleanliness conditions and providing labeling and supplementary instructions for the proper clinical administration of CSPs.
Compounding supervisors shall ensure, through either direct measurement or appropriate information sources, that specific CSPs maintain their labeled strength within monograph limits for USP articles, or within 10% if not specified, until their BUDs. All CSPs are prepared in a manner that maintains sterility and minimizes the introduction of particulate matter.
A written quality assurance procedure includes the following in-process checks that are applied, as appropriate, to specific CSPs: accuracy and precision of measuring and weighing; the requirement for sterility; methods of sterilization and purification; safe limits and ranges for strength of ingredients, bacterial endotoxins, and particulate matter; pH; labeling accuracy and completeness; BUD assignment; and packaging and storage requirements. The dispenser shall, when appropriate and practicable, obtain and evaluate results of testing for identity, strength, purity, and sterility before a CSP is dispensed. Qualified licensed healthcare professionals who supervise compounding and dispensing of CSPs shall ensure that the following objectives are achieved:
-
Compounding personnel are adequately skilled, educated, instructed, and trained to correctly perform and document the following activities in their sterile compounding duties:
- perform antiseptic hand cleansing and disinfection of nonsterile compounding surfaces;
- select and appropriately don protective garb;
- maintain or achieve sterility of CSPs in ISO Class 5 (see Table 1) PEC devices and protect personnel and compounding environments from contamination by radioactive, cytotoxic, and chemotoxic drugs (see Hazardous Drugs as CSPs and Radiopharmaceuticals as CSPs);
- identify, weigh, and measure ingredients; and
- manipulate sterile products aseptically, sterilize high-risk level CSPs, and label and quality inspect CSPs.
- Ingredients have their correct identity, quality, and purity.
- Opened or partially used packages of ingredients for subsequent use in CSPs are properly stored under restricted access conditions in the compounding facility. Such packages cannot be used when visual inspection detects unauthorized breaks in the container, closure, and seal; when the contents do not possess the expected appearance, aroma, and texture; when the contents do not pass identification tests specified by the compounding facility; and when either the BUD or expiration date has been exceeded.
- Water-containing CSPs that are nonsterile during any phase of the compounding procedure are sterilized within 6 hours after completing the preparation in order to minimize the generation of bacterial endotoxins.
- Sterilization methods achieve sterility of CSPs while maintaining the labeled strength of active ingredients and the physical integrity of packaging.
- Measuring, mixing, sterilizing, and purifying devices are clean, appropriately accurate, and effective for their intended use.
- Potential harm from added substances and differences in rate and extent of bioavailability of active ingredients for other than oral route of administration are carefully evaluated before such CSPs are dispensed and administered.
- Packaging selected for CSPs is appropriate to preserve the sterility and strength until the BUD.
- While being used, the compounding environment maintains the sterility or the presterilization purity, whichever is appropriate, of the CSP.
-
Labels on CSPs list the names and amounts or concentrations of active ingredients, and the labels or labeling of injections (see Preservation, Packaging, Storage, and Labeling in the General Notices and Requirements) list the names and amounts or concentrations of all ingredients (see Injections 1). Before being dispensed or administered, the clarity of solutions is visually confirmed; also, the identity and amounts of ingredients, procedures to prepare and sterilize CSPs, and specific release criteria are reviewed to ensure their accuracy and completeness.
-
BUDs are assigned on the basis of direct testing or extrapolation from reliable literature sources and other documentation (see Stability Criteria and Beyond-Use Dating under Pharmaceutical Compounding—Nonsterile Preparations 795).
- Procedures for measuring, mixing, dilution, purification, sterilization, packaging, and labeling conform to the correct sequence and quality established for the specified CSP.
- Deficiencies in compounding, labeling, packaging, and quality testing and inspection can be rapidly identified and corrected.
- When time and personnel availability so permit, compounding manipulations and procedures are separated from postcompounding quality inspection and review before CSPs are dispensed.
This chapter emphasizes the need to maintain high standards for the quality and control of processes, components, and environments and for the skill and knowledge of personnel who prepare CSPs. The rigor of in-process quality-control checks and of postcompounding quality inspection and testing increases with the potential hazard of the route of administration. For example, nonsterility, excessive bacterial endotoxin contamination, large errors in strength of correct ingredients, and incorrect ingredients in CSPs are potentially more dangerous to patients when the CSPs are administered into the vascular and central nervous systems than when administered by most other routes.
CSP MICROBIAL CONTAMINATION RISK LEVELS
The three contamination categories for CSPs described in this section are assigned primarily according to the potential for microbial contamination during the compounding of low-risk level CSPs and medium-risk level CSPs or the potential for not sterilizing high-risk level CSPs, any of which would subject patients to risk of harm, including death. High-risk level CSPs must be sterilized before being administered to patients. The appropriate risk level—low, medium, or high—is assigned according to the corresponding probability of contaminating a CSP with (1) microbial contamination (e.g., microbial organisms, spores, endotoxins) and (2) chemical and physical contamination (e.g., foreign chemicals, physical matter). Potential sources of contamination include, but are not limited to, solid and liquid matter from compounding personnel and objects; nonsterile components employed and incorporated before terminal sterilization; inappropriate conditions within the restricted compounding environment; prolonged presterilization procedures with aqueous preparations; and nonsterile dosage forms used to compound CSPs.
The characteristics described below for low-, medium-, and high-risk level CSPs are intended as a guide to the breadth and depth of care necessary in compounding, but they are neither exhaustive nor prescriptive. The licensed healthcare professionals who supervise compounding are responsible for determining the procedural and environmental quality practices and attributes that are necessary for the risk level they assign to specific CSPs.
These risk levels apply to the quality of CSPs immediately after the final aseptic mixing or filling or immediately after the final sterilization, unless precluded by the specific characteristics of the preparation. Upon subsequent storage and shipping of freshly finished CSPs, an increase in the risks of chemical degradation of ingredients, contamination from physical damage to packaging, and permeability of plastic and elastomeric packaging is expected. In such cases, compounding personnel are responsible for considering the potential additional risks to the integrity of CSPs when assigning BUDs. The pre-administration storage duration and temperature limits specified in the following subsections apply in the absence of direct sterility testing results that justify different limits for specific CSPs.
Low-Risk Level CSPs
CSPs compounded under all the following conditions are at a low risk of contamination.
Low-Risk Conditions—
- The CSPs are compounded with aseptic manipulations entirely within ISO Class 5 (see Table 1) or better air quality using only sterile ingredients, products, components, and devices.
- The compounding involves only transfer, measuring, and mixing manipulations using not more than three commercially manufactured packages of sterile products and not more than two entries into any one sterile container or package (e.g., bag, vial) of sterile product or administration container/device to prepare the CSP.
- Manipulations are limited to aseptically opening ampuls, penetrating disinfected stoppers on vials with sterile needles and syringes, and transferring sterile liquids in sterile syringes to sterile administration devices, package containers of other sterile products, and containers for storage and dispensing.
-
For a low-risk level preparation, in the absence of passing a sterility test (see Sterility Tests 71), the storage periods cannot exceed the following time periods: before administration, the CSPs are properly stored and are exposed for not more than 48 hours at controlled room temperature (see General Notices and Requirements), for not more than 14 days at a cold temperature (see General Notices and Requirements), and for 45 days in solid frozen state between 25
 and 10.
and 10.
Examples of Low-Risk Compounding—
- Single-volume transfers of sterile dosage forms from ampuls, bottles, bags, and vials using sterile syringes with sterile needles, other administration devices, and other sterile containers. The solution content of ampuls should be passed through a sterile filter to remove any particles.
- Simple aseptic measuring and transferring with not more than three packages of manufactured sterile products, including an infusion or diluent solution to compound drug admixtures and nutritional solutions.
Low-Risk Level CSPs with 12-Hour or Less BUD—
If the PEC is a CAI or CACI that does not meet the requirements described in Placement of Primary Engineering Controls or is a laminar airflow workbench (LAFW) or a biological safety cabinet (BSC) that cannot be located within an ISO Class 7 (see Table 1) buffer area, then only low-risk level nonhazardous and radiopharmaceutical CSPs pursuant to a physician's order for a specific patient may be prepared, and administration of such CSPs shall commence within 12 hours of preparation or as recommended in the manufacturers' package insert, whichever is less. Low-risk level CSPs with a 12-hour or less BUD shall meet all of the following four criteria:
- PECs (LAFWs, BSCs, CAIs, CACIs,) shall be certified and maintain ISO Class 5 (see Table 1) as described in Facility Design and Environmental Controls for exposure of critical sites and shall be in a segregated compounding area restricted to sterile compounding activities that minimize the risk of CSP contamination.
- The segregated compounding area shall not be in a location that has unsealed windows or doors that connect to the outdoors or high traffic flow, or that is adjacent to construction sites, warehouses, or food preparation. Note that this list is not intended to be all inclusive.
- Personnel shall follow the procedures described in Personnel Cleansing and Garbing and Additional Personnel Requirements prior to compounding. Sinks should not be located adjacent to the ISO Class 5 (see Table 1) PEC. Sinks should be separated from the immediate area of the ISO Class 5 (see Table 1) PEC device.
- The specifications in Cleaning and Disinfecting the Sterile Compounding Areas, Personnel Training and Competency Evaluation of Garbing, Aseptic Work Practices and Cleaning/Disinfection Procedures, and Viable and Nonviable Environmental Sampling (ES) Testing shall be followed as described in the chapter.
Compounding personnel must recognize that the absence of an ISO Class 7 (see Table 1) buffer area environment in a general uncontrolled environment increases the potential of microbial contamination, and administration durations of microbially contaminated CSPs exceeding a few hours increase the potential for clinically significant microbial colonization, and thus for patient harm, especially in critically ill or immunocompromised patients.
Quality Assurance—
Quality assurance practices include, but are not limited to the following:
- Routine disinfection and air quality testing of the direct compounding environment to minimize microbial surface contamination and maintain ISO Class 5 (see Table 1) air quality.
- Visual confirmation that compounding personnel are properly donning and wearing appropriate items and types of protective garments, including eye protection and face masks.
- Review of all orders and packages of ingredients to ensure that the correct identity and amounts of ingredients were compounded.
- Visual inspection of CSPs to ensure the absence of particulate matter in solutions, the absence of leakage from vials and bags, and the accuracy and thoroughness of labeling.
Media-Fill Test Procedure—
This test or an equivalent test is performed at least annually by each person authorized to compound in a low-risk level environment under conditions that closely simulate the most challenging or stressful conditions encountered during compounding of low-risk level CSPs. Once begun, this test is completed without interruption. Example of test procedure: within an ISO Class 5 (see Table 1) air quality environment, three sets of four 5-mL aliquots of sterile Soybean–Casein Digest Medium (also known as trypticase soy broth or trypticase soy agar [TSA]) are transferred with the same sterile 10-mL syringe and vented needle combination into separate sealed, empty, sterile 30-mL clear vials (i.e., four 5-mL aliquots into each of three 30-mL vials). Sterile adhesive seals are aseptically affixed to the rubber closures on the three filled vials, then the vials are incubated at 20 to 25 or at 30 to 35 for a minimum of 14 days. If two temperatures are used for incubation of media-filled samples, then these filled containers should be incubated for at least 7 days at each temperature (see Microbiological Evaluation of Clean Rooms and Other Controlled Environments 1116). Inspect for microbial growth over 14 days as described in Personnel Training and Competency Evaluation of Garbing, Aseptic Work Practices and Cleaning/Disinfection Procedures.
Medium-Risk Level CSPs
When CSPs are compounded aseptically under Low-Risk Conditions and one or more of the following conditions exists, such CSPs are at a medium risk of contamination.
Medium-Risk Conditions—
- Multiple individual or small doses of sterile products are combined or pooled to prepare a CSP that will be administered either to multiple patients or to one patient on multiple occasions.
- The compounding process includes complex aseptic manipulations other than the single-volume transfer.
- The compounding process requires unusually long duration, such as that required to complete dissolution or homogeneous mixing.
-
For a medium-risk preparation, in the absence of passing a sterility test (see Sterility Tests 71), the storage periods cannot exceed the following time periods: before administration, the CSPs are properly stored and are exposed for not more than 30 hours at controlled room temperature (see General Notices and Requirements), for not more than 9 days at a cold temperature (see General Notices and Requirements), and for 45 days in solid frozen state between 25 and 10.
Examples of Medium-Risk Compounding—
- Compounding of total parenteral nutrition fluids using manual or automated devices during which there are multiple injections, detachments, and attachments of nutrient source products to the device or machine to deliver all nutritional components to a final sterile container.
- Filling of reservoirs of injection and infusion devices with more than three sterile drug products and evacuation of air from those reservoirs before the filled device is dispensed.
- Transfer of volumes from multiple ampuls or vials into one or more final sterile containers.
Quality Assurance—
Quality assurance procedures for medium-risk level CSPs include all those for low-risk level CSPs, as well as a more challenging media-fill test passed annually or more frequently.
Media-Fill Test Procedure—
This test or an equivalent test is performed at least annually under conditions that closely simulate the most challenging or stressful conditions encountered during compounding. Once begun, this test is completed without interruption. Example of test procedure: within an ISO Class 5 (see Table 1) air quality environment, six 100-mL aliquots of sterile Soybean–Casein Digest Medium are aseptically transferred by gravity through separate tubing sets into separate evacuated sterile containers. The six containers are then arranged as three pairs, and a sterile 10-mL syringe and 18-gauge needle combination is used to exchange two 5-mL aliquots of medium from one container to the other container in the pair. For example, after a 5-mL aliquot from the first container is added to the second container in the pair, the second container is agitated for 10 seconds, then a 5-mL aliquot is removed and returned to the first container in the pair. The first container is then agitated for 10 seconds, and the next 5-mL aliquot is transferred from it back to the second container in the pair. Following the two 5-mL aliquot exchanges in each pair of containers, a 5-mL aliquot of medium from each container is aseptically injected into a sealed, empty, sterile 10-mL clear vial, using a sterile 10-mL syringe and vented needle. Sterile adhesive seals are aseptically affixed to the rubber closures on the three filled vials, then the vials are incubated at 20 to 25 or at 30 to 35 for a minimum of 14 days. If two temperatures are used for incubation of media-filled samples, then these filled containers should be incubated for at least 7 days at each temperature (see Microbiological Evaluation of Clean Rooms and Other Controlled Environments 1116). Inspect for microbial growth over 14 days as described in Personnel Training and Competency Evaluation of Garbing, Aseptic Work Practices and Cleaning/Disinfection Procedures.
High-Risk Level CSPs
CSPs compounded under any of the following conditions are either contaminated or at a high risk to become contaminated.
High-Risk Conditions—
- Nonsterile ingredients, including manufactured products not intended for sterile routes of administration (e.g., oral), are incorporated or a nonsterile device is employed before terminal sterilization.
-
Any of the following are exposed to air quality worse than ISO Class 5 (see Table 1) for more than 1 hour (see Immediate-Use CSPs):
- sterile contents of commercially manufactured products,
- CSPs that lack effective antimicrobial preservatives, and
- sterile surfaces of devices and containers for the preparation, transfer, sterilization, and packaging of CSPs.
- Compounding personnel are improperly garbed and gloved (see Personnel Cleansing and Use of Barrier Protective Equipment).
- Nonsterile water-containing preparations are stored for more than 6 hours before being sterilized.
-
It is assumed, and not verified by examination of labeling and documentation from suppliers or by direct determination, that the chemical purity and content strength of ingredients meet their original or compendial specifications in unopened or in opened packages of bulk ingredients (see Ingredient Selection under Pharmaceutical Compounding—Nonsterile Preparations 795).
For a sterilized high-risk level preparation, in the absence of passing a sterility test, the storage periods cannot exceed the following time periods: before administration, the CSPs are properly stored and are exposed for not more than 24 hours at controlled room temperature (see General Notices and Requirements), for not more than 3 days at a cold temperature (see General Notices and Requirements), and for 45 days in solid frozen state between 25 and 10. [note—Sterility tests for autoclaved CSPs are not required unless they are prepared in batches of more than 25 units.]
All nonsterile measuring, mixing, and purifying devices are rinsed thoroughly with sterile, pyrogen-free water, and then thoroughly drained or dried immediately before use for high-risk compounding. All high-risk level CSP solutions subjected to terminal sterilization are prefiltered by passing through a filter with a nominal pore size not larger than 1.2 µm preceding or during filling into their final containers to remove particulate matter. Sterilization of high-risk level CSPs by filtration shall be performed with a sterile 0.2-µm or 0.22-µm nominal pore size filter entirely within an ISO Class 5 (see Table 1) or superior air quality environment.
Examples of High-Risk Conditions—
- Dissolving nonsterile bulk drug and nutrient powders to make solutions that will be terminally sterilized.
- Exposing the sterile ingredients and components used to prepare and package CSPs to room air quality worse than ISO Class 5 (see Table 1) for more than 1 hour (see Immediate-Use CSPs).
- Measuring and mixing sterile ingredients in nonsterile devices before sterilization is performed.
- Assuming, without appropriate evidence or direct determination, that packages of bulk ingredients contain at least 95% by weight of their active chemical moiety and have not been contaminated or adulterated between uses.
Quality Assurance—
Quality assurance procedures for high-risk level CSPs include all those for low-risk level CSPs. In addition, a media-fill test that represents high-risk level compounding is performed semiannually by each person authorized to compound high-risk level CSPs.
Media-Fill Test Procedure for CSPs Sterilized by Filtration—
This test or an equivalent test is performed under conditions that closely simulate the most challenging or stressful conditions encountered when compounding high-risk level CSPs. Once begun, this test is completed without interruption. Example of test procedure (in the following sequence):
- Dissolve 3 g of nonsterile commercially available Soybean–Casein Digest Medium in 100 mL of nonbacteriostatic water to make a 3% nonsterile solution.
- Draw 25 mL of the medium into each of three 30-mL sterile syringes. Transfer 5 mL from each syringe into separate sterile 10-mL vials. These vials are the positive controls to generate exponential microbial growth, which is indicated by visible turbidity upon incubation.
-
Under aseptic conditions and using aseptic techniques, affix a sterile 0.2-µm or 0.22-µm nominal pore size filter unit and a 20-gauge needle to each syringe. Inject the next 10 mL from each syringe into three separate 10-mL sterile vials. Repeat the process for three more vials. Label all vials, affix sterile adhesive seals to the closure of the nine vials, and incubate them at 20 to 25 or at 30 to 35 for a minimum of 14 days. If two temperatures are used for incubation of media-filled samples, then these filled containers should be incubated for at least 7 days at each temperature (see Microbiological Evaluation of Clean Rooms and Other Controlled Environments 1116). Inspect for microbial growth over 14 days as described in Personnel Training and Competency Evaluation of Garbing, Aseptic Work Practices and Cleaning/Disinfection Procedures.
PERSONNEL TRAINING AND EVALUATION IN ASEPTIC MANIPULATION SKILLS
Personnel who prepare CSPs shall be trained conscientiously and skillfully by expert personnel and through audio–video instructional sources and professional publications in the theoretical principles and practical skills of aseptic manipulations and in achieving and maintaining ISO Class 5 (see Table 1) environmental conditions before they begin to prepare CSPs. Compounding personnel shall perform didactic review and pass written and media-fill testing of aseptic manipulative skills initially, at least annually thereafter for low- and medium-risk level compounding, and semiannually for high-risk level compounding. Compounding personnel who fail written tests or whose media-fill test vials result in gross microbial colonization shall be immediately re-instructed and re-evaluated by expert compounding personnel to ensure correction of all aseptic practice deficiencies.
Media-Fill Challenge Testing—
The skill of personnel to aseptically prepare CSPs may be evaluated using sterile fluid bacterial culture media-fill verification3 (i.e., sterile bacterial culture medium transfer via a sterile syringe and needle). Media-fill testing is used to assess the quality of the aseptic skill of compounding personnel. Media-fill tests represent the most challenging or stressful conditions actually encountered by the personnel being evaluated when they prepare particular risk level CSPs and when sterilizing high-risk level CSPs. Media-fill challenge tests that simulate high-risk level compounding are also used to verify the capability of the compounding environment and process to produce a sterile preparation.
Commercially available sterile fluid culture media, such as Soybean–Casein Digest Medium (see Sterility Tests 71), shall be able to promote exponential colonization of bacteria that are most likely to be transmitted to CSPs from the compounding personnel and environment. Media-filled vials are generally incubated at 20 to 25 or at 30 to 35 for a minimum of 14 days. If two temperatures are used for incubation of media-filled samples, then these filled containers should be incubated for at least 7 days at each temperature (see Microbiological Evaluation of Clean Rooms and Other Controlled Environments 1116). Failure is indicated by visible turbidity in the medium on or before 14 days.
IMMEDIATE-USE CSPs
The immediate-use provision is intended only for those situations where there is a need for emergency or immediate patient administration of a CSP. Such situations may include cardiopulmonary resuscitation, emergency room treatment, preparation of diagnostic agents, or critical therapy where the preparation of the CSP under conditions described for Low-Risk Level CSPs subjects the patient to additional risk due to delays in therapy. Immediate-use CSPs are not intended for storage for anticipated needs or batch compounding. Preparations that are medium-risk level and high-risk level CSPs shall not be prepared as immediate-use CSPs.
Immediate-use CSPs are exempt from the requirements described for Low-Risk Level CSPs only when all of the following criteria are met:
- The compounding process involves simple transfer of not more than three commercially manufactured packages of sterile nonhazardous products or diagnostic radiopharmaceutical products from the manufacturers' original containers and not more than two entries into any one container or package (e.g., bag, vial) of sterile infusion solution or administration container/device. For example, anti-neoplastics shall not be prepared as immediate-use CSPs because they are hazardous drugs.
- Unless required for the preparation, the compounding procedure is a continuous process not to exceed 1 hour.
- During preparation, aseptic technique is followed and, if not immediately administered, the finished CSP is under continuous supervision to minimize the potential for contact with nonsterile surfaces, introduction of particulate matter or biological fluids, mix-ups with other CSPs, and direct contact of outside surfaces.
- Administration begins not later than 1 hour following the start of the preparation of the CSP.
- Unless immediately and completely administered by the person who prepared it or immediate and complete administration is witnessed by the preparer, the CSP shall bear a label listing patient identification information, the names and amounts of all ingredients, the name or initials of the person who prepared the CSP, and the exact 1-hour BUD and time.
- If administration has not begun within 1 hour following the start of preparing the CSP, the CSP shall be promptly, properly, and safely discarded.
Compounding in worse than ISO Class 5 (see Table 1) conditions increases the likelihood of microbial contamination, and administration durations of microbially contaminated CSPs exceeding a few hours increase the potential for clinically significant microbial colonization and thus for patient harm, especially in critically ill or immunocompromised patients.
SINGLE-DOSE AND MULTIPLE-DOSE CONTAINERS
Opened or needle-punctured single-dose containers, such as bags, bottles, syringes, and vials of sterile products and CSPs shall be used within 1 hour if opened in worse than ISO Class 5 (see Table 1) air quality (see Immediate-Use CSPs), and any remaining contents must be discarded. Single-dose vials exposed to ISO Class 5 (see Table 1) or cleaner air may be used up to 6 hours after initial needle puncture. Opened single-dose ampuls shall not be stored for any time period. Multiple-dose containers (e.g., vials) are formulated for removal of portions on multiple occasions because they usually contain antimicrobial preservatives. The BUD after initially entering or opening (e.g., needle-punctured) multiple-dose containers is 28 days (see Antimicrobial Effectiveness Testing 51) unless otherwise specified by the manufacturer.
HAZARDOUS DRUGS AS CSPs
Although the potential therapeutic benefits of compounded sterile hazardous drug preparations generally outweigh the risks of their adverse effects in ill patients, exposed healthcare workers risk similar adverse effects with no therapeutic benefit. Occupational exposure to hazardous drugs can result in (1) acute effects, such as skin rashes; (2) chronic effects, including adverse reproductive events; and (3) possibly cancer (see Appendix A of NIOSH Publication no. 2004-165).
Hazardous drugs shall be prepared for administration only under conditions that protect the healthcare workers and other personnel in the preparation and storage areas. Hazardous drugs shall be stored separately from other inventory in a manner to prevent contamination and personnel exposure. Many hazardous drugs have sufficient vapor pressures that allow volatilization at room temperature; thus storage is preferably within a containment area such as a negative pressure room. The storage area should have sufficient general exhaust ventilation, at least 12 air changes per hour (ACPH)4 to dilute and remove any airborne contaminants.
Hazardous drugs shall be handled with caution at all times using appropriate chemotherapy gloves during receiving, distribution, stocking, inventorying, preparation for administration, and disposal. Hazardous drugs shall be prepared in an ISO Class 5 (see Table 1) environment with protective engineering controls in place and following aseptic practices specified for the appropriate contamination risk levels defined in this chapter. Access shall be limited to areas where drugs are stored and prepared to protect persons not involved in drug preparation.
All hazardous drugs shall be prepared in a BSC5 or a CACI that meets or exceeds the standards for CACI in this chapter. The ISO Class 5 (see Table 1) BSC or CACI shall be placed in an ISO Class 7 (see Table 1) area that is physically separated (i.e., a different area from other preparation areas) and optimally has not less than 0.01-inch water column negative pressure to adjacent positive pressure ISO Class 7 (see Table 1) or better ante-areas, thus providing inward airflow to contain any airborne drug. A pressure indicator shall be installed that can be readily monitored for correct room pressurization. The BSC and CACI optimally should be 100% vented to the outside air through HEPA filtration.
If a CACI that meets the requirements of this chapter is used outside of a buffer area, the compounding area shall maintain a minimum negative pressure of 0.01-inch water column and have a minimum of 12 ACPHs.
When closed-system vial-transfer devices (CSTDs) (i.e., vial-transfer systems that allow no venting or exposure of hazardous substance to the environment) are used, they shall be used within the ISO Class 5 (see Table 1) environment of a BSC or CACI. The use of a CSTD is preferred because of their inherent closed system process. In facilities that prepare a low volume of hazardous drugs, the use of two tiers of containment (e.g., CSTD within a BSC or CACI that is located in a non-negative pressure room) is acceptable.
Appropriate personnel protective equipment (PPE) shall be worn when compounding in a BSC or CACI and when using CSTD devices. PPE should include gowns, face masks, eye protection, hair covers, shoe covers or dedicated shoes, double gloving with sterile chemo-type gloves, and compliance with manufacturers' recommendations when using a CACI.
All personnel who compound hazardous drugs shall be fully trained in the storage, handling, and disposal of these drugs. This training shall occur prior to preparing or handling hazardous CSPs, and its effectiveness shall be verified by testing specific hazardous drugs preparation techniques. Such verification shall be documented for each person at least annually. This training shall include didactic overview of hazardous drugs, including mutagenic, teratogenic, and carcinogenic properties, and it shall include ongoing training for each new hazardous drug that enters the marketplace. Compounding personnel of reproductive capability shall confirm in writing that they understand the risks of handling hazardous drugs. The training shall include at least the following: (1) safe aseptic manipulation practices; (2) negative pressure techniques when utilizing a BSC or CACI; (3) correct use of CSTD devices; (4) containment, cleanup, and disposal procedures for breakages and spills; and (5) treatment of personnel contact and inhalation exposure.
note—Because standards of assay and unacceptable quantities of contamination of each drug have not been established in the literature, the following paragraph is a recommendation only. Future standards will be adopted as these assay methods are developed and proven.
In order to ensure containment, especially in operations preparing large volumes of hazardous drugs, environmental sampling to detect uncontained hazardous drugs should be performed routinely (e.g., initially as a benchmark and at least every 6 months or more often as needed to verify containment). This sampling should include surface wipe sampling of the working area of BSCs and CACIs; counter tops where finished preparations are placed; areas adjacent to BSCs and CACIs, including the floor directly under the working area; and patient administration areas. Common marker hazardous drugs that can be assayed include cyclophosphamide, ifosfamide, methotrexate, and fluorouracil. If any measurable contamination (cyclophosphamide levels greater than 1.00 ng per cm2 have been found to cause human uptake) is found by any of these quality assurance procedures, practitioners shall make the decision to identify, document, and contain the cause of contamination. Such action may include retraining, thorough cleaning (utilizing high-pH soap and water), and improving engineering controls. Examples of improving engineering controls are (1) venting BSCs or CACIs 100% to the outside, (2) implementing a CSTD, or (3) re-assessing types of BSCs or CACIs.
Disposal of all hazardous drug wastes shall comply with all applicable federal and state regulations. All personnel who perform routine custodial waste removal and cleaning activities in storage and preparation areas for hazardous drugs shall be trained in appropriate procedures to protect themselves and prevent contamination.
RADIOPHARMACEUTICALS AS CSPs
In the case of production of radiopharmaceuticals for positron emission tomography (PET), general test chapter Radiopharmaceuticals for Positron Emission Tomography—Compounding 823 supersedes this chapter. Upon release of a PET radiopharmaceutical as a finished drug product from a production facility, the further handling, manipulation, or use of the product will be considered compounding, and the content of this section and chapter is applicable.
For the purposes of this chapter, radiopharmaceuticals compounded from sterile components in closed sterile containers and with a volume of 100 mL or less for a single-dose injection or not more than 30 mL taken from a multiple-dose container (see Injections 1) shall be designated as, and conform to, the standards for Low-Risk Level CSPs.
These radiopharmaceuticals shall be compounded using appropriately shielded vials and syringes in a properly functioning and certified ISO Class 5 (see Table 1) PEC located in an ISO Class 8 (see Table 1) or cleaner air environment to permit compliance with special handling, shielding, and negative air flow requirements.
Radiopharmaceutical vials designed for multi-use, compounded with technetium-99m, exposed to ISO Class 5 (see Table 1) environment, and punctured by needles with no direct contact contamination may be used up to the time indicated by manufacturers' recommendations. Storage and transport of properly shielded vials of radiopharmaceutical CSPs may occur in a limited access ambient environment without a specific ISO class designation.
Technetium-99m/molybdenum-99 generator systems shall be stored and eluted (operated) under conditions recommended by manufacturers and applicable state and federal regulations. Such generator systems shall be eluted in an ISO Class 8 (see Table 1) or cleaner air environment to permit special handling, shielding, and air flow requirements. To limit acute and chronic radiation exposure of inspecting personnel to a level that is as low as reasonably achievable (ALARA), direct visual inspection of radiopharmaceutical CSPs containing high concentrations of doses of radioactivity shall be conducted in accordance with ALARA.
Radiopharmaceuticals prepared as Low-Risk Level CSPs with 12-Hour or Less BUD shall be prepared in a segregated compounding area. A line of demarcation defining the segregated compounding area shall be established. Materials and garb exposed in a patient care and treatment area shall not cross a line of demarcation into the segregated compounding area.
ALLERGEN EXTRACTS AS CSPs
Allergen extracts as CSPs are single-dose and multiple-dose intradermal or subcutaneous injections that are prepared by specially trained physicians and personnel under their direct supervision. Allergen extracts as CSPs are not subject to the personnel, environmental, and storage requirements for all CSP Microbial Contamination Risk Levels in this chapter only when all of the following criteria are met:
- The compounding process involves simple transfer via sterile needles and syringes of commercial sterile allergen products and appropriate sterile added substances (e.g., glycerin, phenol in sodium chloride injection).
- All allergen extracts as CSPs shall contain appropriate substances in effective concentrations to prevent the growth of microorganisms. Nonpreserved allergen extracts shall comply with the appropriate CSP risk level requirements in the chapter.
- Before beginning compounding activities, personnel perform a thorough hand-cleansing procedure by removing debris from under fingernails using a nail cleaner under running warm water followed by vigorous hand and arm washing to the elbows for at least 30 seconds with either nonantimicrobial or antimicrobial soap and water.
- Compounding personnel don hair covers, facial hair covers, gowns, and face masks.
- Compounding personnel perform antiseptic hand cleansing with an alcohol-based surgical hand scrub with persistent activity.
- Compounding personnel don powder-free sterile gloves that are compatible with sterile 70% isopropyl alcohol (IPA) before beginning compounding manipulations.
- Compounding personnel disinfect their gloves intermittently with sterile 70% IPA when preparing multiple allergen extracts as CSPs.
- Ampul necks and vial stoppers on packages of manufactured sterile ingredients are disinfected by careful wiping with sterile 70% IPA swabs to ensure that the critical sites are wet for at least 10 seconds and allowed to dry before they are used to compound allergen extracts as CSPs.
- The aseptic compounding manipulations minimize direct contact contamination (e.g., from glove fingertips, blood, nasal and oral secretions, shed skin and cosmetics, other nonsterile materials) of critical sites (e.g., needles, opened ampuls, vial stoppers).
- The label of each multiple-dose vial (MDV) of allergen extracts as CSPs lists the name of one specific patient and a BUD and storage temperature range that is assigned based on manufacturers' recommendations or peer-reviewed publications.
- Single-dose allergen extracts as CSPs shall not be stored for subsequent additional use.
Personnel who compound allergen extracts as CSPs must be aware of greater potential risk of microbial and foreign material contamination when allergen extracts as CSPs are compounded in compliance with the foregoing criteria instead of the more rigorous standards in this chapter for CSP Microbial Contamination Risk Levels. Although contaminated allergen extracts as CSPs can pose health risks to patients when they are injected intradermally or subcutaneously, these risks are substantially greater if the extract is inadvertently injected intravenously.
VERIFICATION OF COMPOUNDING ACCURACY AND STERILITY
The compounding procedures and sterilization methods for CSPs correspond to correctly designed and verified written documentation in the compounding facility. Verification requires planned testing, monitoring, and documentation to demonstrate adherence to environmental quality requirements, personnel practices, and procedures critical to achieving and maintaining sterility, accuracy, and purity of finished CSPs. For example, sterility testing (see Test for Sterility of the Product To Be Examined under Sterility Tests 71) may be applied to specimens of low- and medium-risk level CSPs, and standard self-contained biological indicators (BI) shall be added to nondispensable specimens of high-risk level CSPs before terminal sterilization for subsequent evaluation to determine whether the sterilization cycle was adequate (see Biological Indicators for Sterilization 1035). Packaged and labeled CSPs shall be visually inspected for physical integrity and expected appearance, including final fill amount. The accuracy of identities, concentrations, amounts, and purities of ingredients in CSPs shall be confirmed by reviewing labels on packages, observing and documenting correct measurements with approved and correctly standardized devices, and reviewing information in labeling and certificates of analysis provided by suppliers. When the correct identity, purity, strength, and sterility of ingredients and components of CSPs cannot be confirmed (in cases of, for example, unlabeled syringes, opened ampuls, punctured stoppers of vials and bags, containers of ingredients with incomplete labeling), such ingredients and components shall be discarded immediately.
Some individual ingredients, such as bulk drug substances, are not labeled with expiration dates when they are stable indefinitely in their commercial packages under their labeled storage conditions. However, despite retaining full chemical stability, such ingredients may gain or lose moisture during storage and use. Changes in moisture content may require testing (see Loss on Drying 731) to determine the correct amount to weigh for accurate content of active chemical moieties in CSPs (see Pharmaceutical Calculations in Prescription Compounding 1160).
Although not required, a quantitative stability-indicating chemical assay is recommended to ensure compounding accuracy of CSPs, especially those that contain drug ingredients with a narrow therapeutic plasma concentration range.
Sterilization Methods
The licensed healthcare professionals who supervise compounding shall be responsible for determining that the selected sterilization method (see Methods of Sterilization under Sterilization and Sterility Assurance of Compendial Articles 1211) both sterilizes and maintains the strength, purity, quality, and packaging integrity of CSPs. The selected sterilization process is obtained from experience and appropriate information sources (e.g., see Sterilization and Sterility Assurance of Compendial Articles 1211)—and, preferably, verified wherever possible—to achieve sterility in the particular CSPs. General guidelines for matching CSPs and components to appropriate sterilization methods include the following:
- CSPs have been ascertained to remain physically and chemically stable when subjected to the selected sterilization method.
-
Glass and metal devices may be covered tightly with aluminum foil, then exposed to dry heat in an oven at a mean temperature of 250 for 30 minutes to achieve sterility and depyrogenation (see Dry-Heat Sterilization under Sterilization and Sterility Assurance of Compendial Articles 1211 and Bacterial Endotoxins Test 85). Such items are either used immediately or stored until use in an environment suitable for compounding Low-Risk Level CSPs and Medium-Risk Level CSPs.
- Personnel ascertain from appropriate information sources that the sterile microporous membrane filter used to sterilize CSP solutions, during either compounding or administration, is chemically and physically compatible with the CSP.
sterilization of high-risk level csps by filtration
Commercially available sterile filters shall be approved for human-use applications in sterilizing pharmaceutical fluids. Sterile filters used to sterilize CSPs shall be pyrogen free and have a nominal pore size of 0.2 or 0.22 µm. They shall be certified by the manufacturer to retain at least 107 microorganisms of a strain of Brevundimonas (Pseudomonas) diminuta on each square centimeter of upstream filter surface area under conditions similar to those in which the CSPs will be sterilized (see High-Risk Conditions in High-Risk Level CSPs).
The compounding supervisor shall ensure, directly or from appropriate documentation, that the filters are chemically and physically stable at the pressure and temperature conditions to be used, that they have enough capacity to filter the required volumes, and that they will achieve sterility and maintain prefiltration pharmaceutical quality, including strength of ingredients of the specific CSP. The filter dimensions and liquid material to be sterile-filtered shall permit the sterilization process to be completed rapidly, without the replacement of the filter during the process. When CSPs are known to contain excessive particulate matter, a prefilter of larger nominal pore size membrane is placed upstream from the sterilizing filter to remove gross particulate contaminants in order to maximize the efficiency of the sterilizing filter.
Filter units used to sterilize CSPs shall also be subjected to manufacturers' recommended integrity test, such as the bubble point test.
Compounding personnel shall ascertain that selected filters will achieve sterilization of the particular CSPs being sterilized. Large deviations from usual or expected chemical and physical properties of CSPs (e.g., water-miscible alcohols) may cause undetectable damage to filter integrity and shrinkage of microorganisms to sizes smaller than filter nominal pore size.
sterilization of high-risk level csps by steam
The process of thermal sterilization employing saturated steam under pressure, or autoclaving, is the preferred method to terminally sterilize aqueous preparations that have been verified to maintain their full chemical and physical stability under the conditions employed (see Steam Sterilization under Sterilization and Sterility Assurance of Compendial Articles 1211). To achieve sterility, all materials are to be exposed to steam at 121 under a pressure of about 1 atmosphere or 15 psi for the duration verified by testing to achieve sterility of the items, which is usually 20 to 60 minutes for CSPs. An allowance shall be made for the time required for the material to reach 121 before the sterilization exposure duration is timed.
Not directly exposing items to pressurized steam may result in survival of microbial organisms and spores. Before their sterilization, plastic, glass, and metal devices are tightly wrapped in low-particle-shedding paper or fabrics or sealed in envelopes that prevent poststerilization microbial penetration. Immediately before filling ampuls and vials that will be steam sterilized, solutions are passed through a filter having a nominal pore size not larger than 1.2 µm for removal of particulate matter. Sealed containers shall be able to generate steam internally; thus, stoppered and crimped empty vials shall contain a small amount of moisture to generate steam.
The description of steam sterilization conditions and duration for specific CSPs shall be included in written documentation in the compounding facility. The effectiveness of steam sterilization shall be verified using appropriate BIs of Bacillus stearothermophilus (see Biological Indicators 1035) and other confirmation methods such as temperature-sensing devices (see Sterilization and Sterility Assurance of Compendial Articles 1211 and Sterility Tests 71).
sterilization of high-risk level csps by dry heat
Dry heat sterilization is usually done as a batch process in an oven designed for sterilization. Heated filtered air shall be evenly distributed throughout the chamber by a blower device. The oven should be equipped with a system for controlling temperature and exposure period. Sterilization by dry heat requires higher temperatures and longer exposure times than does sterilization by steam. Dry heat shall be used only for those materials that cannot be sterilized by steam, when either the moisture would damage the material or the material is impermeable. During sterilization, sufficient space shall be left between materials to allow for good circulation of the hot air. The description of dry heat sterilization conditions and duration for specific CSPs shall be included in written documentation in the compounding facility. The effectiveness of dry heat sterilization shall be verified using appropriate BIs of Bacillus subtilis (see Biological Indicators 1035) and other confirmation methods such as temperature-sensing devices (see Sterilization and Sterility Assurance of Compendial Articles 1211 and Sterility Tests 71). [note—Dry heat sterilization may be performed at a lower temperature than may be effective for depyrogenation].
Depyrogenation by Dry Heat
Dry heat depyrogenation shall be used to render glassware or containers such as vials free from pyrogens as well as viable microbes. A typical cycle would be 30 minutes at 250. The description of the dry heat depyrogenation cycle and duration for specific load items shall be included in written documentation in the compounding facility. The effectiveness of the dry heat depyrogenation cycle shall be verified using endotoxin challenge vials (ECVs). The bacterial endotoxin test should be performed on the ECVs to verify that the cycle is capable of achieving a 3-log reduction in endotoxin (see Sterilization and Sterility Assurance of Compendial Articles 1211 and Bacterial Endotoxins Test 85).
ENVIRONMENTAL QUALITY AND CONTROL
Achieving and maintaining sterility and overall freedom from contamination of a CSP is dependent on the quality status of the components incorporated, the process utilized, personnel performance, and the environmental conditions under which the process is performed. The standards required for the environmental conditions depend on the amount of exposure of the CSP to the immediate environment anticipated during processing. The quality and control of environmental conditions for each risk level of operation are explained in this section. In addition, operations using nonsterile components require the use of a method of preparation designed to produce a sterile preparation.
Exposure of Critical Sites
Maintaining the sterility and cleanliness (i.e., freedom from sterile foreign materials) of critical sites is a primary safeguard for CSPs. Critical sites are locations that include any component or fluid pathway surfaces (e.g., vial septa, injection ports, beakers) or openings (e.g., opened ampuls, needle hubs) exposed and at risk of direct contact with air (e.g., ambient room or HEPA filtered), moisture (e.g., oral and mucosal secretions), or touch contamination. The risk of, or potential for, critical sites to be contaminated with microorganisms and foreign matter increases with increasing exposed area of the critical sites, the density or concentration of contaminants, and exposure duration to worse than ISO Class 5 (see Table 1) air. Examples include an opened ampul or vial stopper on a 10-mL or larger vial or an injection port on a package of intravenous solution having an area larger than the point of a needle or the tip of a syringe.
The nature of a critical site also affects the risk of contamination. The relatively rough, permeable surface of an elastomeric closure retains microorganisms and other contaminants after swabbing with a sterile 70% IPA pad more readily than does the smoother glass surface of the neck of an ampul. Therefore, the surface disinfection can be expected to be more effective for an ampul.
Protection of critical sites by precluding physical contact and airborne contamination shall be given the highest priority in sterile compounding practice. Airborne contaminants, especially those generated by sterile compounding personnel, are much more likely to reach critical sites than are contaminants that are adhering to the floor or other surfaces below the work level. Furthermore, large and high-density particles that are generated and introduced by compounding manipulations and personnel have the potential to settle on critical sites even when those critical sites are exposed within ISO Class 5 (see Table 1) air.
ISO Class 5 Air Sources, Buffer Areas, and Ante-Areas
The most common sources of ISO Class 5 (see Table 1) air quality for exposure of critical sites are horizontal and vertical LAFWs, CAIs, and CACIs. A clean room (see Microbiological Evaluation of Clean Rooms and Other Controlled Environments 1116) is a compounding environment that is supplied with HEPA or HEPA-filtered air that meets ISO Class 7 (see Table 1), the access to which is limited to personnel trained and authorized to perform sterile compounding and facility cleaning. A buffer area is an area that provides at least ISO Class 7 (see Table 1) air quality.
Figure 1 is a conceptual representation of the placement of an ISO Class 5 (see Table 1) PEC in a segregated compounding area used for low-risk level CSPs with 12-hour or less BUD. This plan depicts the most critical operation area located within the PEC in a designated area (see definition of Segregated Compounding Area) separated from activities not essential to the preparation of CSPs. Placement of devices (e.g., computers, printers) and objects (e.g., carts, cabinets) that are not essential to compounding in the segregated area should be restricted or limited, depending on their effect on air quality in the ISO Class 5 (see Table 1) PEC.
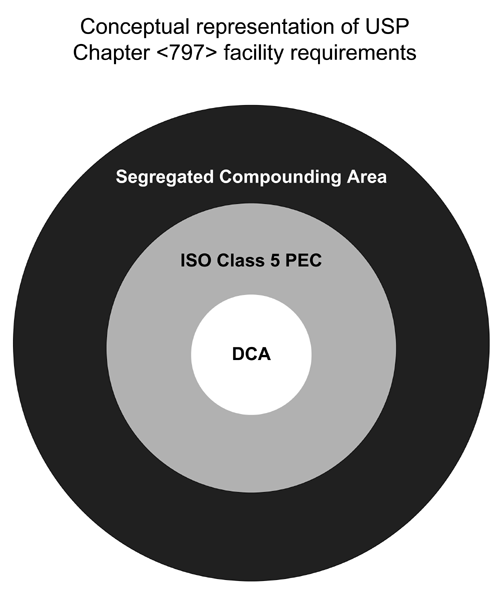
Figure 1. Conceptual representation of the placement of an ISO Class 5 PEC in a segregated compounding area used for low-risk level CSPs with 12-hour or less BUD.
Figure 2 is a conceptual representation of the arrangement of a facility for preparation of CSPs categorized as low-, medium-, and high-risk level. The quality of the environmental air increases with movement from the outer boundary to the direct compounding area (DCA). Placement of devices in ante-areas and buffer areas is dictated by their effect on the designated environmental quality of atmospheres and surfaces, which shall be verified by monitoring (see Viable and Nonviable Environmental Sampling (ES) Testing). It is the responsibility of each compounding facility to ensure that each source of ISO Class 5 (see Table 1) environment for exposure of critical sites and sterilization by filtration is properly located, operated, maintained, monitored, and verified.

Figure 2. Conceptual representation of the arrangement of a facility for preparation of CSPs categorized as low-, medium-, and high-risk level.
Placement of devices (e.g., computers, printers) and objects (e.g., carts, cabinets) that are not essential to compounding in buffer areas is dictated by their effect on the required environmental quality of air atmospheres and surfaces, which shall be verified by monitoring (see Viable and Nonviable Environmental Sampling (ES) Testing). It is the responsibility of each compounding facility to ensure that each source of ISO Class 5 (see Table 1) environment for exposure of critical sites and sterilization by filtration is properly located, operated, maintained, monitored, and verified.
Facility Design and Environmental Controls
Compounding facilities are physically designed and environmentally controlled to minimize airborne contamination from contacting critical sites. These facilities shall also provide a comfortable and well-lighted working environment, which typically includes a temperature of 20 or cooler, to maintain comfortable conditions for compounding personnel to perform flawlessly when attired in the required aseptic compounding garb. PECs typically include, but are not limited to, LAFWs, BSCs, CAIs, and CACIs, which provide an ISO Class 5 (see Table 1) environment for the exposure of critical sites. PECs shall maintain ISO Class 5 (see Table 1) or better conditions for 0.5-µm particles (dynamic operating conditions) while compounding CSPs. Secondary engineering controls such as buffer areas and ante-areas generally serve as a core for the location of the PEC. Buffer areas are designed to maintain at least ISO Class 7 (see Table 1) conditions for 0.5-µm particles under dynamic conditions and ISO Class 8 (see Table 1) conditions for 0.5-µm and larger particles under dynamic conditions for the ante-areas. Airborne contamination control is achieved in the PEC through the use of HEPA filters. The airflow in the PEC shall be unidirectional (laminar flow), and because of the particle collection efficiency of the filter, the “first air” at the face of the filter is, for the purposes of aseptic compounding, free from airborne particulate contamination. HEPA-filtered air shall be supplied in critical areas (ISO Class 5, see Table 1) at a velocity sufficient to sweep particles away from the compounding area and maintain unidirectional airflow during operations. Proper design and control prevents turbulence and stagnant air in the critical area. In situ air pattern analysis via smoke studies shall be conducted at the critical area to demonstrate unidirectional airflow and sweeping action over and away from the product under dynamic conditions.
The principles of HEPA-filtered unidirectional airflow in the work environment shall be understood and practiced in the compounding process in order to achieve the desired environmental conditions. Policies and procedures for maintaining and working within the PEC area shall be written and followed. The policies and procedures will be determined by the scope and risk levels of the aseptic compounding activities utilized during the preparation of the CSPs. The CSP work environment is designed to have the cleanest work surfaces (PEC) located in a buffer area. The buffer area shall maintain at least ISO Class 7 (see Table 1) conditions for 0.5-µm and larger particles under dynamic operating conditions. The room shall be segregated from surrounding, unclassified spaces to reduce the risk of contaminants being blown, dragged, or otherwise introduced into the filtered unidirectional airflow environment, and this segregation shall be continuously monitored. For rooms providing a physical separation through the use of walls, doors, and pass-throughs, a minimum differential positive pressure of 0.02- to 0.05-inch water column is required. For buffer areas not physically separated from the ante-areas, the principle of displacement airflow shall be employed. This concept utilizes a low pressure differential, high airflow principle. Using displacement airflow typically requires an air velocity of 40 ft per minute or more from the buffer area across the line of demarcation into the ante-area.
The displacement concept shall not be used for high-risk compounding.6 The PEC shall be placed within a buffer area in such a manner as to avoid conditions that could adversely affect their operation. For example, strong air currents from opened doors, personnel traffic, or air streams from the HVAC systems can disrupt the unidirectional airflow in open-faced workbenches. The operators may also create disruptions in airflow by their own movements and by the placement of objects onto the work surface. The PEC shall be placed out of the traffic flow and in a manner to avoid disruption from the HVAC system and room cross-drafts. Room air exchanges are typically expressed as ACPHs. Adequate HEPA-filtered airflow supplied to the buffer area and ante-area is required to maintain cleanliness classification during operational activity through the number of ACPHs. Factors that should be considered when determining air-change requirements include number of personnel working in the room and compounding processes that generate particulates, as well as temperature effects. An ISO Class 7 (see Table 1) buffer area and ante-area supplied with HEPA-filtered air shall receive an ACPH of not less than 30. The PEC is a good augmentation to generating air changes in the air supply of an area but cannot be the sole source of HEPA-filtered air. If the area has an ISO Class 5 (see Table 1) recirculating device, a minimum of 15 ACPHs through the area supply HEPA filters is adequate, providing the combined ACPH is not less than 30. More air changes may be required, depending on the number of personnel and processes. HEPA-filtered supply air shall be introduced at the ceiling, and returns should be mounted low on the wall, creating a general top-down dilution of area air with HEPA-filtered make-up air. Ceiling-mounted returns are not recommended. All HEPA filters should be efficiency tested using the most penetrating particle size and should be leak tested at the factory and then leak tested again in situ after installation.7
Activities and tasks carried out within the buffer area shall be limited to only those necessary when working within a controlled environment. Only the furniture, equipment, supplies, and other material required for the compounding activities to be performed shall be brought into the area, and they shall be nonpermeable, nonshedding, cleanable, and resistant to disinfectants. Whenever such items are brought into the area, they shall first be cleaned and disinfected. Whenever possible, equipment and other items used in the buffer area shall not be taken out of the area except for calibration, servicing, or other activities associated with the proper maintenance of the item.
The surfaces of ceilings, walls, floors, fixtures, shelving, counters, and cabinets in the buffer area shall be smooth, impervious, free from cracks and crevices, and nonshedding, thereby promoting cleanability and minimizing spaces in which microorganisms and other contaminants may accumulate. The surfaces shall be resistant to damage by disinfectant agents. Junctures of ceilings to walls shall be coved or caulked to avoid cracks and crevices where dirt can accumulate. If ceilings consist of inlaid panels, the panels shall be impregnated with a polymer to render them impervious and hydrophobic, and they shall be caulked around each perimeter to seal them to the support frame. Walls may be constructed of flexible material (e.g., heavy gauge polymer), panels locked together and sealed, or of epoxy-coated gypsum board. Preferably, floors are overlaid with wide sheet vinyl flooring with heat-welded seams and coving to the sidewall. Dust-collecting overhangs, such as ceiling utility pipes, and ledges, such as windowsills, should be avoided. The exterior lens surface of ceiling lighting fixtures should be smooth, mounted flush, and sealed. Any other penetrations through the ceiling or walls shall be sealed. The buffer area shall not contain sources of water (sinks) or floor drains. Work surfaces shall be constructed of smooth, impervious materials, such as stainless steel or molded plastic, so that they are easily cleaned and disinfected. Carts should be of stainless steel wire, nonporous plastic, or sheet metal construction with good quality, cleanable casters to promote mobility. Storage shelving, counters, and cabinets shall be smooth, impervious, free from cracks and crevices, nonshedding, cleanable, and disinfectable; their number, design, and manner of installation shall promote effective cleaning and disinfection.
Placement of Primary Engineering Controls
- Only authorized personnel and materials required for compounding and cleaning shall be permitted in the buffer area.
- Presterilization procedures for high-risk level CSPs, such as weighing and mixing, shall be completed in no worse than an ISO Class 8 (see Table 1) environment.
- PECs shall be located out of traffic patterns and away from room air currents that could disrupt the intended airflow patterns.
CAIs and CACIs shall be placed in an ISO Class 7 (see Table 1) buffer area unless they meet all of the following conditions:
- The isolator shall provide isolation from the room and maintain ISO Class 5 (see Table 1) during dynamic operating conditions, including transferring ingredients, components, and devices into and out of the isolator and during preparation of CSPs.
- Particle counts sampled approximately 6 to 12 inches upstream of the critical exposure site shall maintain ISO Class 5 (see Table 1) levels during compounding operations.
- Not more than 3520 particles (0.5 µm and larger) per m3 shall be counted during material transfer, with the particle counter probe located as near to the transfer door as possible without obstructing the transfer.8
It is incumbent on the compounding personnel to obtain documentation from the manufacturer that the CAI/CACI will meet this standard when located in environments where the background particle counts exceed ISO Class 8 (see Table 1) for 0.5-µm and larger particles. When isolators are used for sterile compounding, the recovery time to achieve ISO Class 5 (see Table 1) air quality shall be documented and internal procedures developed to ensure that adequate recovery time is allowed after material transfer before and during compounding operations.
If the PEC is a CAI or CACI that does not meet the requirements above or is a LAFW or BSC that cannot be located within an ISO Class 7 (see Table 1) buffer area, then only low-risk level nonhazardous and radiopharmaceutical CSPs pursuant to a physician order for a specific patient may be prepared, and administration of the CSP shall commence within 12 hours of preparation or as recommended in the manufacturer's package insert, whichever is less.
Viable and Nonviable Environmental Sampling (ES) Testing
The ES program should provide information to staff and leadership to demonstrate that the PEC is maintaining an environment within the compounding area that consistently ensures acceptably low viable and nonviable particle levels. The compounding area includes the ISO Class 5 (see Table 1) PEC (LAFWs, BSCs, CAIs, and CACIs), buffer areas, ante-areas, and segregated compounding areas.
Environmental sampling shall occur as part a comprehensive quality management program and shall occur minimally under any of the following conditions:
- as part of the commissioning and certification of new facilities and equipment;
- following any servicing of facilities and equipment;
- as part of the re-certification of facilities and equipment (i.e., every 6 months);
- in response to identified problems with end products or staff technique; or
- in response to issues with CSPs, observed compounding personnel work practices, or patient-related infections (where the CSP is being considered as a potential source of the infection).
environmental nonviable particle testing program
A program to sample nonviable airborne particles differs from that for viable particles in that it is intended to directly measure the performance of the engineering controls used to create the various levels of air cleanliness, for example, ISO Class 5, 7, or 8 (see Table 1).
Engineering Control Performance Verification—
PECs (LAFWs, BSCs, CAIs, and CACIs) and secondary engineering controls (buffer and ante-areas) are essential components of the overall contamination control strategy for aseptic compounding. As such, it is imperative that they perform as designed and that the resulting levels of contamination be within acceptable limits. Certification procedures such as those outlined in Certification Guide for Sterile Compounding Facilities (CAG-003-2006)9 shall be performed by a qualified individual no less than every 6 months and whenever the device or room is relocated or altered or major service to the facility is performed.
Total Particle Counts—
Certification that each ISO classified area, for example, ISO Class 5, 7, and 8 (see Table 1), is within established guidelines shall be performed no less than every 6 months and whenever the LAFW, BSC, CAI, or CACI is relocated or the physical structure of the buffer area or ante-area has been altered. Testing shall be performed by qualified operators using current, state-of-the-art electronic equipment with results of the following:
- ISO Class 5: not more than 3520 particles 0.5 µm and larger size per cubic meter of air for any LAFW, BSC, CAI, and CACI;
- ISO Class 7: not more than 352,000 particles of 0.5 µm size and larger per cubic meter of air for any buffer area;
- ISO Class 8: not more than 3,520,000 particles or 0.5 µm size and larger per cubic meter of air for any ante-area.
All certification records shall be maintained and reviewed by supervising personnel or other designated employees to ensure that the controlled environments comply with the proper air cleanliness, room pressures, and ACPHs.
pressure differential monitoring
A pressure gauge or velocity meter shall be installed to monitor the pressure differential or airflow between the buffer area and the ante-area and between the ante-area and the general environment outside the compounding area. The results shall be reviewed and documented on a log at least every work shift (minimum frequency shall be at least daily) or by a continuous recording device. The pressure between the ISO Class 7 (see Table 1) and the general pharmacy area shall not be less than 5 Pa (0.02 inch water column). In facilities where low- and medium-risk level CSPs are prepared, differential airflow shall maintain a minimum velocity of 0.2 meters per second (40 feet per minute) between buffer area and ante-area.
environmental viable airborne particle testing program
The risk of contaminating a CSP prepared under low-risk level and medium-risk level conditions is highly dependent on proper hand hygiene and garbing practices, compounding personnel aseptic technique, and the presence of surface contamination, assuming that all work is performed in a certified and properly functioning ISO Class 5 (see Table 1) PEC and secondary engineering controls, ISO Class 7 (see Table 1) buffer area, and ISO Class 8 (see Table 1) ante-area. High-risk level CSPs pose the greatest threat to patients because compounding personnel are tasked with the requirement of processing nonsterile components and devices in order to achieve sterility.
A sampling program in conjunction with an observational audit is designed to evaluate the competency of compounding personnel work practices, allowing for the implementation of corrective actions on an ongoing basis (see Personnel Training and Competency Evaluation of Garbing, Aseptic Work Practices and Cleaning/Disinfection Procedures).
Sampling Plan—
An appropriate environmental sampling plan shall be developed for airborne viable particles based on a risk assessment of compounding activities performed.
Selected sampling sites shall include locations within each ISO Class 5 (see Table 1) environment and in the ISO Class 7 and 8 (see Table 1) areas and in the segregated compounding areas at greatest risk of contamination (e.g., work areas near the ISO Class 5 [see Table 1] environment, counters near doors, pass-through boxes). The plan shall include sample location, method of collection, frequency of sampling, volume of air sampled, and time of day as related to activity in the compounding area and action levels.
Review of the data generated during a sampling event may detect elevated amounts of airborne microbial bioburden; such changes may be indicative of adverse changes within the environment. It is recommended that compounding personnel refer to Microbiological Evaluation of Clean Rooms and Other Controlled Environments 1116 and the CDC's “Guidelines for Environmental Infection Control in Healthcare Facilities, 2003” for more information.
Growth Medium—
A general microbiological growth medium such as Soybean–Casein Digest Medium shall be used to support the growth of bacteria. Malt extract agar or some other media that support the growth of fungi shall be used in high-risk level compounding environments. Media used for surface sampling must be supplemented with additives to neutralize the effects of disinfecting agents (e.g., TSA with lecithin and polysorbate 80).
Viable Air Sampling—
Evaluation of airborne microorganisms using volumetric collection methods in the controlled air environments (LAFWs, CAIs, clean room or buffer areas, and ante-areas) shall be performed by properly trained individuals for all compounding risk levels.
Impaction shall be the preferred method of volumetric air sampling. Use of settling plates for qualitative air sampling may not be able to determine adequately the quality of air in the controlled environment. The settling of particles by gravity onto culture plates depends on the particle size and may be influenced by air movement. Consequently, the number of colony-forming units (cfu) on a settling plate may not always relate to the concentrations of viable particles in the sampled environment.
For low-, medium-, and high-risk level compounding, air sampling shall be performed at locations that are prone to contamination during compounding activities and during other activities such as staging, labeling, gowning, and cleaning. Locations shall include zones of air backwash turbulence within LAFW and other areas where air backwash turbulence may enter the compounding area (doorways, in and around ISO Class 5 [see Table 1] PEC and environments). Consideration should be given to the overall effect the chosen sampling method will have on the unidirectional airflow within a compounding environment.
For low-risk level CSPs with 12-hour or less BUD prepared in a PEC (LAFWs, BSCs, CAIs) that maintains an ISO Class 5 (see Table 1), air sampling shall be performed at locations inside the ISO Class 5 (see Table 1) environment and other areas that are in close proximity to the ISO Class 5 (see Table 1) environment during the certification of the PEC.
Air Sampling Devices—
There are a number of manufacturers of electronic air sampling equipment. It is important that personnel refer to the manufacturer's recommended procedures when using the equipment to perform volumetric air sampling procedures. The instructions in the manufacturer's user's manual for verification and use of electric air samplers that actively collect volumes of air for evaluation must be followed. A sufficient volume of air (400 to 1000 liters) shall be tested at each location in order to maximize sensitivity. The volumetric air sampling devices need to be serviced and calibrated as recommended by the manufacturer.
It is recommended that compounding personnel also refer to Methodology and Instrumentation for Quantitation of Viable Airborne Microorganisms under Microbiological Evaluation of Clean Rooms and Other Controlled Environments 1116, which provides more information on the use of volumetric air samplers and volume of air that should be sampled to detect environmental bioburden excursions.
Air Sampling Frequency and Process—
Air sampling shall be performed at least semiannually (i.e., every 6 months) as part of the re-certification of facilities and equipment. If compounding occurs in multiple locations within an institution (e.g., main pharmacy, satellites), environmental sampling is required for each individual compounding area. A sufficient volume of air shall be sampled and the manufacturer's guidelines for use of the electronic air sampling equipment followed. Any facility construction or equipment servicing may require that air sampling be performed during these events.
Incubation Period—
At the end of the designated sampling or exposure period for air sampling activities, the microbial growth media plates are recovered and their covers secured (e.g., taped), and they are inverted and incubated at a temperature and for a time period conducive to multiplication of microorganisms. TSA should be incubated at 30 to 35 for 48 to 72 hours. Malt extract agar or other suitable fungal media should be incubated at 26 to 30 for 5 to 7 days. The number of discrete colonies of microorganisms are counted and reported as cfu and documented on an environmental sampling form. Counts from air sampling need to be transformed into cfu per cubic meter of air and evaluated for adverse trends.
Action Levels, Documentation, and Data Evaluation—
The value of viable microbial sampling of the air in the compounding environment is realized when the data are used to identify and correct an unacceptable situation. Sampling data shall be collected and reviewed on a periodic basis as a means of evaluating the overall control of the compounding environment. If an activity consistently shows elevated levels of microbial growth, competent microbiology personnel shall be consulted.
Any cfu count that exceeds its respective action level (see Table 2) should prompt a re-evaluation of the adequacy of personnel work practices, cleaning procedures, operational procedures, and air filtration efficiency within the aseptic compounding location. An investigation into the source of the contamination shall be conducted. Sources could include HVAC systems, damaged HEPA filters, and changes in personnel garbing or work practices. The source of the problem shall be eliminated, the affected area cleaned, and resampling performed.
Counts of cfu are to be used as an approximate measure of the environmental microbial bioburden. Action levels are determined on the basis of cfu data gathered at each sampling location and trended over time. The numbers in Table 2 should be used only as guidelines. Regardless of the number of cfu identified in the pharmacy, further corrective actions will be dictated by the identification of microorganisms recovered (at least the genus level) by an appropriate credentialed laboratory of any microbial bioburden captured as a cfu using an impaction air sampler. Highly pathogenic microorganisms (e.g., Gram-negative rods, coagulase positive staphylococcus, molds and yeasts) can be potentially fatal to patients receiving CSPs and must be immediately remedied, regardless of cfu count, with the assistance of a competent microbiologist, infection control professional, or industrial hygienist.
Table 2. Recommended Action Levels for Microbial Contamination*
†(cfu per cubic meter [1000 liters] of air per plate)
†(cfu per cubic meter [1000 liters] of air per plate)
| Classification | Air Sample† |
| ISO Class 5 | > 1 |
| ISO Class 7 | > 10 |
| ISO Class 8 or worse | > 100 |
|
*
Guidance for Industry–Sterile Drug Products Produced by Aseptic Processing–Current Good Manufacturing Practice–US HHS, FDA September 2004.
|
|
Additional Personnel Requirements
Food, drinks, and materials exposed in patient care and treatment areas shall not enter ante-areas, buffer areas, or segregated compounding areas where components and ingredients of CSPs are present. When compounding activities require the manipulation of a patient's blood-derived or other biological material (e.g., radiolabeling a patient's or donor's white blood cells), the manipulations shall be clearly separated from routine material-handling procedures and equipment used in CSP preparation activities, and they shall be controlled by specific SOPs in order to avoid any cross-contamination. Packaged compounding supplies and components, such as needles, syringes, tubing sets, and small- and large-volume parenterals, should be uncartoned and wiped down with a disinfectant that does not leave a residue (e.g., sterile 70% IPA), when possible in an ante-area of ISO Class 8 (see Table 1) air quality, before being passed into the buffer areas. Personnel hand hygiene and garbing procedures are also performed in the ante-area, which may contain a sink that enables hands-free use with a closed system of soap dispensing to minimize the risk of extrinsic contamination. There shall be some demarcation designation that separates the ante-area from the buffer area. Adequate provision for performing antiseptic hand cleansing using an alcohol-based surgical hand scrub with persistent activity followed by the donning of sterile gloves should be provided after entry into the buffer area.
Cleaning and Disinfecting the Compounding Area
Environmental contact is a major source of microbial contamination of CSPs. Consequently, scrupulous attention to cleaning and disinfecting the sterile compounding areas is required to minimize this as a source of CSP contamination.
The cleaning and disinfecting practices and frequencies in this section apply to ISO Class 5 (see Table 1) compounding areas for exposure of critical sites as well as buffer areas, ante-areas, and segregated compounding areas. Compounding personnel are responsible for ensuring that the frequency of cleaning is in accordance with the requirements stated in Table 3 and determining the cleaning and disinfecting products to be used (see Appendix II). Any organizational or institutional policies regarding disinfectant selection should be considered by compounding personnel. All cleaning and disinfecting practices and policies for the compounding of CSPs shall be included in written SOPs and shall be followed by all compounding personnel.
The selection and use of disinfectants in healthcare facilities is guided by several properties, such as microbicidal activity, inactivation by organic matter, residue, and shelf life (see Appendix II). In general, highly toxic disinfectants, such as glutaraldehyde, are not used on housekeeping surfaces (e.g., floors, countertops). Many disinfectants registered by the EPA are one-step disinfectants. This means that the disinfectant has been formulated to be effective in the presence of light to moderate soiling without a pre-cleaning step.
Surfaces in LAFWs, BSCs, CAIs, and CACIs, which are intimate to the exposure of critical sites, require disinfecting more frequently than do housekeeping surfaces such as walls and ceilings. Disinfecting sterile compounding areas shall occur on a regular basis at the intervals noted in Table 3 when spills occur, when the surfaces are visibly soiled, and when microbial contamination is known to have been or is suspected of having been introduced into the compounding areas.
When the surface to be disinfected has heavy soiling, a cleaning step is recommended prior to the application of the disinfectant. Trained compounding personnel are responsible for developing, implementing, and practicing the procedures for cleaning and disinfecting the DCAs written in the SOPs. Cleaning and disinfecting shall occur before compounding is performed. Items shall be removed from all areas to be cleaned, and surfaces shall be cleaned by removing loose material and residue from spills; for example, water-soluble solid residues are removed with sterile water (for injection or irrigation) and low-shedding wipes. This shall be followed by wiping with a residue-free disinfecting agent such as sterile 70% IPA, which is allowed to dry before compounding begins.
Cleaning and disinfecting surfaces in the LAFWs, BSCs, CAIs, and CACIs are the most critical practices before the preparation of CSPs. Consequently, such surfaces shall be cleaned and disinfected frequently, including at the beginning of each work shift, before each batch preparation is started, every 30 minutes during continuous compounding periods of individual CSPs, when there are spills, and when surface contamination is known or suspected from procedural breaches.
Work surfaces in the ISO Class 7 (see Table 1) buffer areas and ISO Class 8 (see Table 1) ante-areas as well as segregated compounding areas shall be cleaned and disinfected at least daily, and dust and debris shall be removed when necessary from storage sites for compounding ingredients and supplies using a method that does not degrade the ISO Class 7 or 8 (see Table 1) air quality (see Disinfectants and Antiseptics 1072).
Table 3. Minimum Frequency of Cleaning and Disinfecting Compounding Areas
| Site | Minimum Frequency |
| ISO Class 5 (see Table 1) Primary Engineering Control (e.g., LAFW, BSC, CAI, CACI) | At the beginning of each shift, before each batch, not longer than 30 minutes following the previous surface disinfection when ongoing compounding activities are occurring, after spills, and when surface contamination is known or suspected |
| Counters and easily cleanable work surfaces | Daily |
| Floors | Daily |
| Walls | Monthly |
| Ceilings | Monthly |
| Storage shelving | Monthly |
Floors in the buffer or clean area, ante-area, and segregated compounding area are cleaned by mopping with a cleaning and disinfecting agent once daily at a time when no aseptic operations are in progress. Mopping shall be performed by trained personnel using approved agents and procedures described in the written SOPs. It is incumbent on compounding personnel to ensure that such cleaning is performed properly. In the buffer or clean area, ante-area, and segregated compounding area, walls, ceilings, and shelving shall be cleaned and disinfected monthly. Cleaning and disinfecting agents are to be used with careful consideration of compatibilities, effectiveness, and inappropriate or toxic residues (see Appendix II). Their schedules of use and methods of application shall be in accordance with written SOPs and followed by custodial or compounding personnel.
All cleaning materials, such as wipers, sponges, and mops, shall be nonshedding, preferably composed of synthetic micro fibers, and dedicated to use in the buffer or clean area, ante-area, and segregated compounding areas and shall not be removed from these areas except for disposal. Floor mops may be used in both the buffer or clean area and ante-area, but only in that order. Ideally, all cleaning tools are discarded after one use by collection in suitable plastic bags and removed with minimal agitation. If cleaning materials (e.g., mops) are reused, procedures shall be developed (based on manufacturers' recommendations) that ensure that the effectiveness of the cleaning device is maintained and that repeated use does not add to the bioburden of the area being cleaned.
Supplies and equipment removed from shipping cartons shall be wiped with a suitable disinfecting agent (e.g., sterile 70% IPA) delivered from a spray bottle or other suitable delivery method. After the disinfectant is sprayed or wiped on a surface to be disinfected, the disinfectant shall be allowed to dry, during which time the item shall not be used for compounding purposes.
Wiping with small sterile 70% IPA swabs that are commercially available in individual foil-sealed packages (or a comparable method) is preferred for disinfecting entry points on bags and vials, allowing the IPA to dry before piercing stoppers with sterile needles and breaking necks of ampuls. The surface of the sterile 70% IPA swabs used for disinfecting entry points of sterile packages and devices shall not contact any other object before contacting the surface of the entry point. Sterile 70% IPA wetted gauze pads or other particle-generating material shall not be used to disinfect the sterile entry points of packages and devices.
When sterile supplies are received in sealed pouches designed to keep them sterile until opening, the sterile supplies may be removed from the covering pouches as the supplies are introduced into the ISO Class 5 (see Table 1) PEC (LAFW, BSC, CAI, CACI) without the need to disinfect the individual sterile supply items. No shipping or other external cartons may be taken into the buffer or clean area or segregated compounding area.
Personnel Cleansing and Garbing
The careful cleansing of hands and arms and the correct donning of PPE by compounding personnel constitute the first major step in preventing microbial contamination in CSPs. Personnel shall also be thoroughly competent and highly motivated to perform flawless aseptic manipulations with ingredients, devices, and components of CSPs. Squamous cells are normally shed from the human body at a rate of 106 or more per hour, and those skin particles are laden with microorganisms.10, 11 When individuals are experiencing rashes, sunburn, weeping sores, conjunctivitis, active respiratory infection, as well as when they wear cosmetics, they shed these particles at even higher rates. Particles shed from compounding personnel pose an increased risk of microbial contamination of critical sites of CSPs. Therefore, compounding personnel with such conditions as mentioned above shall be excluded from working in ISO Class 5 (see Table 1) and ISO Class 7 (see Table 1) compounding areas until their conditions are remedied.
Before entering the buffer area or segregated compounding area (see Low-Risk Level CSPs with 12-Hour or Less BUD), compounding personnel shall remove personal outer garments (e.g., bandannas, coats, hats, jackets, scarves, sweaters, vests); all cosmetics, because they shed flakes and particles; and all hand, wrist, and other visible jewelry or piercings (e.g., earrings, lip or eyebrow piercings) that can interfere with the effectiveness of PPE (e.g., fit of gloves and cuffs of sleeves). The wearing of artificial nails or extenders is prohibited while working in the sterile compounding environment. Natural nails shall be kept neat and trimmed.
Personnel shall don the following PPE in an order that proceeds from those activities considered the dirtiest to those considered the cleanest. Garbing activities considered the dirtiest include donning of dedicated shoes or shoe covers, head and facial hair covers (e.g., beard covers in addition to face masks), and face masks/eye shields. Eye shields are optional unless working with irritants such as germicidal disinfecting agents or when preparing hazardous drugs.
After donning dedicated shoes or shoe covers, head and facial hair covers, and face masks, a hand cleansing procedure shall be performed by removing debris from underneath fingernails using a nail cleaner under running warm water followed by vigorous hand washing. Hands and forearms shall be washed to the elbows for at least 30 seconds with soap (either nonantimicrobial or antimicrobial) and water while in the ante-area. The use of antimicrobial scrub brushes is not recommended because they can cause skin irritation and skin damage. Hands and forearms to the elbows will be completely dried using either lint-free disposable towels or an electronic hand dryer. After completion of hand washing, a nonshedding gown with sleeves that fit snugly around the wrists and enclosed at the neck is donned. Gowns designated for buffer area use shall be worn, and preferably they should be disposable. If reusable gowns are worn, they should be laundered appropriately for buffer area use.
Once inside the buffer area or segregated compounding area (see Low-Risk Level CSPs with 12-Hour or Less BUD), and prior to donning sterile powder-free gloves, antiseptic hand cleansing shall be performed using a waterless alcohol-based surgical hand scrub with persistent activity12 following manufacturers' recommendations. Hands are allowed to dry thoroughly before donning sterile gloves.
Sterile gloves shall be the last item donned before compounding begins. Gloves become contaminated when they contact nonsterile surfaces during compounding activities. Disinfection of contaminated gloved hands may be accomplished by wiping or rubbing sterile 70% IPA to all contact surface areas of the gloves and letting the gloved hands dry thoroughly. Only use gloves that have been tested for compatibility with alcohol disinfection by the manufacturer. Routine application of sterile 70% IPA shall occur throughout the compounding process and whenever nonsterile surfaces (e.g. vials, counter tops, chairs, carts) are touched. Gloves on hands shall also be routinely inspected for holes, punctures, or tears and replaced immediately if such are detected. Antiseptic hand cleansing shall be performed as indicated above. Compounding personnel shall be trained and evaluated in the avoidance of touching critical sites.
When compounding personnel exit the compounding area during a work shift, the exterior gown may be removed and retained in the compounding area if not visibly soiled, to be re-donned during that same work shift only. However, shoe covers, hair and facial hair covers, face masks/eye shields, and gloves shall be replaced with new ones before re-entering the compounding area, and proper hand hygiene shall be performed.
During high-risk compounding activities that precede terminal sterilization, such as weighing and mixing of nonsterile ingredients, compounding personnel shall be garbed and gloved the same as when performing compounding in an ISO Class 5 (see Table 1) environment. Properly garbed and gloved compounding personnel who are exposed to air quality that is either known or suspected to be worse than ISO Class 7 (see Table 1) shall re-garb PPE along with washing their hands properly, performing antiseptic hand cleansing with a waterless alcohol-based surgical hand scrub, and donning sterile gloves upon re-entering the ISO Class 7 (see Table 1) buffer area. When CAIs and CACIs are the source of the ISO Class 5 (see Table 1) environment, the garbing and gloving requirements for compounding personnel should be as described above, unless the isolator manufacturer can provide written documentation based on validated environmental testing that any component(s) of PPE or personnel cleansing are not required.
Personnel Training and Competency Evaluation of Garbing, Aseptic Work Practices, and Cleaning/Disinfection Procedures
Personnel who prepare CSPs shall be trained conscientiously and skillfully by expert personnel and through multimedia instructional sources and professional publications in the theoretical principles and practical skills of garbing procedures, aseptic work practices, achieving and maintaining ISO Class 5 (see Table 1) environmental conditions, and cleaning and disinfection procedures. This training shall be completed and documented before any compounding personnel begin to prepare CSPs. Compounding personnel shall complete didactic training, pass written competence assessments, undergo skill assessment using observational audit tools, and media-fill testing (see Appendices III–V).
Media-fill testing of aseptic work skills shall be performed initially before beginning to prepare CSPs and at least annually thereafter for low- and medium-risk level compounding and semiannually for high-risk level compounding.
Compounding personnel who fail written tests or observational audits or whose media-fill test vials have one or more units showing visible microbial contamination shall be re-instructed and re-evaluated by expert compounding personnel to ensure correction of all aseptic work practice deficiencies. Compounding personnel shall pass all evaluations prior to resuming compounding of sterile preparations. In addition to didactic evaluation and aseptic media fill, compounding personnel must demonstrate proficiency of proper hand hygiene, garbing, and consistent cleaning procedures.
In the event that cleaning and disinfecting procedures are also performed by other support personnel (e.g., institutional environmental services, housekeeping), thorough training of proper hand hygiene, garbing, and cleaning and disinfection procedures shall be done by a qualified aseptic compounding expert. After completion of training, support personnel shall routinely undergo performance evaluation of proper hand hygiene, garbing, and all applicable cleaning and disinfecting procedures conducted by a qualified aseptic compounding expert.
competency evaluation of garbing and aseptic work practice
The risk of contaminating a CSP prepared under low-risk level and medium-risk level conditions is highly dependent on proper hand hygiene and garbing practices, compounding personnel aseptic technique, and the presence of surface contamination, assuming that all work is performed in a certified and properly functioning ISO Class 5 (see Table 1) PEC and secondary engineering controls, ISO Class 7 (see Table 1) buffer area, and ISO Class 8 (see Table 1) ante-area. High-risk level CSPs pose the greatest threat to patients because compounding personnel are tasked with the requirement of processing nonsterile components and devices in order to achieve sterility. Compounding personnel shall be evaluated initially prior to beginning compounding CSPs and whenever an aseptic media fill is performed using a form such as the Sample Form for Assessing Hand Hygiene and Garbing Related Practices of Compounding Personnel (see Appendix III) and the personnel glove fingertip sampling procedures indicated below.
Aseptic Work Practice Assessment and Evaluation via Personnel Glove Fingertip Sampling—
Sampling of compounding personnel glove fingertips shall be performed for all CSP risk level compounding because direct touch contamination is the most likely source of introducing microorganisms into CSPs prepared by humans. Glove fingertip sampling shall be used to evaluate the competency of personnel in performing hand hygiene and garbing procedures in addition to educating compounding personnel on proper work practices, which include frequent and repeated glove disinfection using sterile 70% IPA during actual compounding of CSPs. All personnel shall demonstrate competency in proper hand hygiene and garbing procedures and in aseptic work practices (e.g., disinfection of component surfaces, routine disinfection of gloved hands).
Sterile contact agar plates shall be used to sample the gloved fingertips of compounding personnel after garbing in order to assess garbing competency and after completing the media-fill preparation (without applying sterile 70% IPA) in order to assess the adequacy of aseptic work practices prior to being initially allowed to prepare CSPs for human use and for more experienced personnel to maintain their qualifications to prepare CSPs for human use.
Garbing And Gloving Competency Evaluation—
Compounding personnel shall be visually observed during the process of performing hand hygiene and garbing procedures (see Personnel Cleansing and Garbing under Personnel Training and Evaluation in Aseptic Manipulation Skills above). The visual observation shall be documented on a form such as the Sample Form for Assessing Hand Hygiene and Garbing Related Practices of Compounding Personnel (see Appendix III) and maintained to provide a permanent record and long-term assessment of personnel competency.
Gloved Fingertip Sampling—
All compounding personnel shall successfully complete an initial competency evaluation and gloved fingertip/thumb sampling procedure (zero cfu) no less than three times before initially being allowed to compound CSPs for human use. Immediately after the compounding employee completes the hand hygiene and garbing procedure (e.g., donning of sterile gloves prior to any disinfection with sterile 70% IPA), the evaluator will collect a gloved fingertip and thumb sample from both hands of the compounding employee onto appropriate agar plates by lightly pressing each fingertip into the agar. The plates will be incubated for the appropriate incubation period and at the appropriate temperature (see Incubation Period). After completing the initial gowning and gloving competency evaluation, re-evaluation of all compounding personnel for this competency shall occur at least annually for personnel who compound low- and medium-risk level CSPs and semi-annually for personnel who compound high-risk level CSPs using one or more sample collections during any media-fill test procedure before they are allowed to continue compounding CSPs for human use.
Immediately prior to sampling, gloves shall not be disinfected with sterile 70% IPA. Disinfecting gloves immediately before sampling will provide false negative results. Plates filled with nutrient agar with neutralizing agents such as lecithin and polysorbate 80 added shall be used when sampling personnel fingertips. Personnel shall “touch” the agar with the fingertips of both hands in separate plates in a manner to create a slight impression in the agar. The sampled gloves shall be immediately discarded and proper hand hygiene performed after sampling. The nutrient agar plates shall be incubated as stated below (see Incubation Period). Results should be reported separately as number of cfu per employee per hand (left hand, right hand). The cfu action level for gloved hands will be based on the total number of cfu on both gloves, not per hand.
Incubation Period—
At the end of the designated sampling period for compounding personnel competency assessment activities (surface or personnel), the agar plates are recovered and covers secured and they are inverted and incubated at a temperature and for a time period conducive to multiplication of microorganisms. TSA with lecithin and polysorbate 80 shall be incubated at 30 to 35 for 48 to 72 hours.
Aseptic Manipulation Competency Evaluation—
After successful completion of an initial Hand Hygiene and Garbing Competency Evaluation, all compounding personnel shall have their aseptic technique and related practice competency evaluated initially during the Media-Fill Test Procedure and subsequent annual or semi-annual Media-Fill Test Procedures. Records of these evaluations will be maintained using a form such as the Sample Form for Assessing Aseptic Technique and Related Practices of Compounding Personnel (see Appendix IV) and maintained to provide a permanent record of and long-term assessment of personnel competency.
Media-Fill Test Procedure—
The skill of personnel to aseptically prepare CSPs shall be evaluated using sterile fluid bacterial culture media-fill verification, (i.e., sterile bacterial culture medium transfer via a sterile syringe and needle). Media-fill testing is used to assess the quality of the aseptic skill of compounding personnel. Media-fill tests shall represent the most challenging or stressful conditions actually encountered by the personnel being evaluated when they prepare low- and medium-risk level CSPs and when sterilizing high-risk level CSPs. Media-fill challenge tests are also used to verify the capability of the compounding environment and processes to produce sterile preparations.
A commercially available sterile fluid culture media, such as Soybean–Casein Digest Medium (see Sterility Tests 71), that is able to promote exponential colonization of bacteria that are most likely to be transmitted to CSPs from the compounding personnel and environment is commonly used. For high-risk level CSPs nonsterile commercially available Soybean–Casein Digest Medium may be used to make a 3% solution. Normal processing steps, including filter sterilization, shall be mimicked. Media-filled vials shall be incubated at 20 to 25 or at 30 to 35 for a minimum of 14 days. If two temperatures are used for incubation of media-filled samples, then these filled containers should be incubated for at least 7 days at each temperature (see Microbiological Evaluation of Clean Rooms and Other Controlled Environments 1116). Failure is indicated by visible turbidity in any one of the media-fill units on or before 14 days. Other methodologies recommended by a competent microbiologist to enhance recovery time and sensitivity to detect microbial contamination may be considered (see CSP Microbial Contamination Risk Levels for examples of media-fill procedures).
surface cleaning and disinfection sampling and assessment
Surface sampling is an important component of the maintenance of a suitable microbially controlled environment for compounding CSPs, especially since transfer of microbial contamination from improperly disinfected work surfaces via inadvertent touch contact by compounding personnel can be a potential source of contamination into CSPs. It is useful for evaluating facility and work surface cleaning and disinfecting procedures and employee competency in work practices such as disinfection of component/vial surface cleaning. Surface sampling shall be performed in all ISO classified areas on a periodic basis. Sampling can be accomplished using contact plates or swabs, and it shall be done at the conclusion of compounding. Locations to be sampled shall be defined in a sample plan or on a form. The size of the plate to be used for each sampled location usually ranges from 24 to 30 cm2. Contact plates are filled with general solid agar growth medium and neutralizing agents above the rim of the plate, and they are used for sampling regular or flat surfaces. Swabs may be used for sampling irregular surfaces, especially for equipment (see Microbiological Evaluation of Clean Rooms and Other Controlled Environments 1116).
Cleaning and Disinfecting Competency Evaluation—
Compounding personnel and other personnel responsible for cleaning shall be visually observed during the process of performing cleaning and disinfecting procedures, during initial personnel training on cleaning procedures, during changes in cleaning staff, and at the completion of any media-fill test procedure (see Cleaning and Disinfecting of Compounding Areas).
The visual observation shall be documented using a form such as the Sample Form for Assessing Cleaning and Disinfection Procedures (see Appendix V) and maintained to provide a permanent record and long-term assessment of personnel competency.
Surface Collection Methods—
To sample surfaces using a contact plate, gently touch the sample area with the agar surface and roll the plate across the surface to be sampled. The contact plate will leave a growth media residue behind; therefore, immediately after sampling with the contact plate, the sampled area shall be thoroughly wiped with a nonshedding wipe soaked in sterile 70% IPA.
If an area is sampled via the swab method, collection of the sample is processed by using appropriate procedures that will result in the surface location equivalent to that of a contact plate. After swabbing the surface to be sampled, swabs are placed in an appropriate diluent; an aliquot is planted on or in the specified nutrient agar. Results should be reported as cfu per unit of surface area.
Action Levels, Documentation, and Data Evaluation
The value of viable microbial monitoring of gloved fingertips and surfaces of components and the compounding environment are realized when the data are used to identify and correct an unacceptable work practice. Sampling data shall be collected and reviewed on a routine basis as a means of evaluating the overall control of the compounding environment. If an activity consistently shows elevated levels of microbial growth, competent microbiology personnel shall be consulted.
Any cfu count that exceeds its respective action level (see Table 4) should prompt a re-evaluation of the adequacy of personnel work practices, cleaning procedures, operational procedures, and air filtration efficiency within the aseptic compounding location. An investigation into the source of the contamination shall be conducted. Sources could include HVAC systems, damaged HEPA filters, and changes in personnel garbing or working practices. The source of the problem shall be eliminated, the affected area cleaned, and resampling performed.
When gloved fingertip sample results exceed action levels after proper incubation, a review of hand hygiene and garbing procedures as well as glove and surface disinfection procedures and work practices shall be performed and documented. Employee training may be required to correct the source of the problem.
Counts of cfu are to be used as an approximate measure of the environmental microbial bioburden. Action levels are determined on the basis of cfu data gathered at each sampling location and trended over time. The numbers in Table 4 should be used only as guidelines. Regardless of the number of cfu identified in the compounding facility, further corrective actions will be dictated by the identification of microorganisms recovered (at least the genus level) by an appropriate credentialed laboratory of any microbial bioburden captured as a cfu using an impaction air sampler. Highly pathogenic microorganisms (e.g., Gram-negative rods, coagulase positive staphylococcus, molds and yeasts) can be potentially fatal to patients receiving CSPs and shall be immediately remedied, regardless of cfu count, with the assistance of a competent microbiologist, infection control professional, or industrial hygienist.
Table 4. Recommended Action Levels for Microbial
Contamination*
Contamination*
| Classification | Fingertip Sample | Surface Sample (Contact Plate) (cfu per plate) |
| ISO Class 5 | > 3 | > 3 |
| ISO Class 7 | N/A | > 5 |
| ISO Class 8 or worse | N/A | > 100 |
|
*
Pharmaceutical Inspection Co-operation Scheme (PIC/S) Guide to Good Manufacturing Practice for Medicinal Products Annexes PE 009-6, 5 April 2007.
|
||
SUGGESTED STANDARD OPERATING PROCEDURES (SOPs)
The compounding facility shall have written, properly approved SOPs designed to ensure the quality of the environment in which a CSP is prepared. The following procedures are recommended:
- Access to the buffer area is restricted to qualified personnel with specific responsibilities or assigned tasks in the compounding area.
- All cartoned supplies are decontaminated in the area by removing them from shipping cartons and wiping or spraying them with a nonresidue-generating disinfecting agent while they are being transferred to a clean and properly disinfected cart or other conveyance for introduction into the buffer area. Manufacturers' directions or published data for minimum contact time will be followed. Individual pouched sterile supplies need not be wiped because the pouches can be removed as these sterile supplies are introduced into the buffer area.
- Supplies that are required frequently or otherwise needed close at hand but not necessarily needed for the scheduled operations of the shift are decontaminated and stored on shelving in the ante-area.
- Carts used to bring supplies from the storeroom cannot be rolled beyond the demarcation line in the ante-area, and carts used in the buffer area cannot be rolled outward beyond the demarcation line unless cleaned and disinfected before returning.
- Generally, supplies required for the scheduled operations of the shift are wiped down with an appropriate disinfecting agent and brought into the buffer area, preferably on one or more movable carts. Supplies that are required for back-up or general support of operations may be stored on the designated shelving in the buffer area, but excessive amounts of supplies are to be avoided.
- Nonessential objects that shed particles shall not be brought into the buffer area, including pencils, cardboard cartons, paper towels, and cotton items (e.g., gauze pads).
- Essential paper-related products (e.g., paper syringe overwraps, work records contained in a protective sleeve) shall be wiped down with an appropriate disinfecting agent prior to being brought into the buffer area.
- Traffic flow in and out of the buffer area shall be minimized.
- Personnel preparing to enter the buffer area shall remove all personal outer garments, cosmetics (because they shed flakes and particles), and all hand, wrist, and other visible jewelry or piercings that can interfere with the effectiveness of PPE.
- Personnel entering the ante-area shall don attire as described in Personnel Cleansing and Garbing and Personnel Training and Competency Evaluation of Garbing, Aseptic Work Practices and Cleaning/Disinfection Procedures.
- Personnel shall then thoroughly wash hands and forearms to the elbow with soap and water for at least 30 seconds. An air dryer or disposable nonshedding towels are used to dry hands and forearms after washing.
- Personnel entering the buffer area shall perform antiseptic hand cleansing prior to donning sterile gloves using a waterless alcohol-based surgical hand scrub with persistent activity.
- Chewing gum, drinks, candy, or food items shall not be brought into the buffer area or ante-area. Materials exposed in patient care and treatment areas shall never be introduced into areas where components and ingredients for CSPs are present.
- At the beginning of each compounding activity session, and whenever liquids are spilled, the surfaces of the direct compounding environment are first cleaned with USP Purified Water to remove water-soluble residues. Immediately thereafter, the same surfaces are disinfected with a nonresidue-generating agent using a nonlinting wipe.
- Primary engineering controls shall be operated continuously during compounding activity. When the blower is turned off and before other personnel enter to perform compounding activities, only one person shall enter the buffer area for the purposes of turning on the blower (for at least 30 minutes) and disinfecting the work surfaces.
- Traffic in the area of the DCA is minimized and controlled.
- Supplies used in the DCA for the planned procedures are accumulated and then decontaminated by wiping or spraying the outer surface with sterile 70% IPA or removing the outer wrap at the edge of the DCA as the item is introduced into the aseptic work area.
- All supply items are arranged in the DCA so as to reduce clutter and provide maximum efficiency and order for the flow of work.
- After proper introduction into the DCA of supply items required for and limited to the assigned operations, they are so arranged that a clear, uninterrupted path of HEPA-filtered air will bathe all critical sites at all times during the planned procedures. That is, no objects may be placed between the first air from HEPA filters and an exposed critical site.
- All procedures are performed in a manner designed to minimize the risk of touch contamination. Gloves are disinfected with adequate frequency with an approved disinfectant such as sterile 70% IPA.
- All rubber stoppers of vials and bottles and the necks of ampuls are disinfected by wiping with sterile 70% IPA and waiting for at least 10 seconds before they are used to prepare CSPs.
- After the preparation of every CSP, the contents of the container are thoroughly mixed and then inspected for the presence of particulate matter, evidence of incompatibility, or other defects.
- After procedures are completed, used syringes, bottles, vials, and other supplies are removed, but with a minimum of exit and re-entry into the DCA so as to minimize the risk of introducing contamination into the aseptic workspace.
ELEMENTS OF QUALITY CONTROL
A written description of specific training and performance evaluation program for individuals involved in the use of aseptic techniques for the preparation of sterile products shall be developed for each site. This program equips personnel with the appropriate knowledge and trains them in the required skills necessary to perform the assigned tasks. Each person assigned to the aseptic area in the preparation of sterile products shall successfully complete specialized training in aseptic techniques and aseptic area practices prior to preparing CSPs (see Personnel Training and Evaluation in Aseptic Manipulation Skills and Personnel Training and Competency Evaluation of Garbing, Aseptic Work Practices and Cleaning/Disinfection Procedures).
Ingredients and Devices
Compounding personnel ascertain that ingredients for CSPs are of the correct identity and appropriate quality using the following information: vendor labels, labeling, certificates of analysis, direct chemical analysis, and knowledge of compounding facility storage conditions.
sterile ingredients and devices
Commercially available sterile drug products, sterile ready-to-use containers, and devices are examples of sterile components. A written procedure for unit-by-unit physical inspection preparatory to use is followed to ensure that these components are sterile, free from defects, and otherwise suitable for their intended use.
nonsterile ingredients and devices
If any nonsterile components, including containers and ingredients, are used to make a CSP, such CSPs must be high risk. Nonsterile active ingredients and added substances or excipients for CSPs should preferably be official USP or NF articles. When nonofficial ingredients are used, they shall be accompanied by certificates of analysis from their suppliers to aid compounding personnel in judging the identity, quality, and purity in relation to the intended use in a particular CSP. Physical inspection of a package of ingredients is necessary in order to detect breaks in the container, looseness in the cap or closure, and deviation from the expected appearance, aroma, and texture of the contents.
Bulk or unformulated drug substances and added substances or excipients shall be stored in tightly closed containers under temperature, humidity, and lighting conditions that are either indicated in official monographs or approved by suppliers. The date of receipt by the compounding facility shall be clearly and indelibly marked on each package of ingredient. After receipt by the compounding facility, packages of ingredients that lack a supplier's expiration date cannot be used after 1 year unless either appropriate inspection or testing indicates that the ingredient has retained its purity and quality for use in CSPs.
Careful consideration and evaluation of nonsterile ingredient sources is especially warranted when the CSP will be administered into the vascular system, central nervous system, or eyes.
Upon receipt of each lot of the bulk drug substance or excipient used for CSPs, the individual compounding the preparation performs a visual inspection of the lot for evidence of deterioration, other types of unacceptable quality, and wrong identification. For bulk drug substances or excipients, visual inspection is performed on a routine basis as described in the written protocol.
Equipment
It is necessary that equipment, apparatus, and devices used to compound a CSP be consistently capable of operating properly and within acceptable tolerance limits. Written procedures outlining required equipment calibration, annual maintenance, monitoring for proper function, and controlled procedures for use of the equipment and specified time frames for these activities are established and followed. Routine maintenance and frequencies shall be outlined in these SOPs. Results from the equipment calibration, annual maintenance reports, and routine maintenance are kept on file for the lifetime of the equipment. Personnel are prepared through an appropriate combination of specific training and experience to operate or manipulate any piece of equipment, apparatus, or device they may use when preparing CSPs. Training includes gaining the ability to determine whether any item of equipment is operating properly or is malfunctioning.
VERIFICATION OF AUTOMATED COMPOUNDING DEVICES (ACDs) FOR PARENTERAL NUTRITION COMPOUNDING
ACDs for the preparation of parenteral nutrition admixtures are widely used by pharmacists in hospitals and other healthcare settings. They are designed to streamline the labor-intensive processes involved in the compounding of these multiple-component formulations by automatically delivering the individual nutritional components in a predetermined sequence under computerized control. Parenteral nutrition admixtures often contain 20 or more individual additives representing as many as 50 or more individual components (e.g., 15 to 20 crystalline amino acids, dextrose monohydrate, and lipids; 10 to 12 electrolyte salts; 5 to 7 trace minerals; and 12 vitamins). Thus, ACDs can provide improved accuracy and precision of the compounding process over the traditional manual compounding methods.
Accuracy
The accuracy of an ACD can be determined in various ways to ensure that the correct quantities of nutrients, electrolytes, or other nutritional components are delivered to the final infusion container. Initially, the ACD is tested for its volume and weight accuracy. For volume accuracy, a suitable volume of Sterile Water for Injection, USP, which represents a typical additive volume (e.g., 40 mL for small-volume range of 1 to 100 mL, 300 mL for large-volume range of 100 to 1000 mL), is programmed into the ACD and delivered to the appropriate volumetric container. The compounding personnel should then consult Volumetric Apparatus 31 for appropriate parameters to assess the volumetric performance of the ACD. For gravimetric accuracy, the balance used in conjunction with the ACD is tested using various weight sizes that represent the amounts typically used to deliver the various additives. Compounding personnel should consult Weights and Balances 41 for acceptable tolerances of the weights used. In addition, the same volume of Sterile Water for Injection used to assess volumetric accuracy is then weighed on the balance used in conjunction with the ACD. For example, if 40 mL of water was used in the volumetric assessment, its corresponding weight should be about 40 g (assuming the relative density of water is 1.0). In addition, during the use of the ACD, certain additives, such as potassium chloride (corrected for density differences), can also be tested in the same manner as with an in-process test.
Finally, additional tests of accuracy may be employed that determine the content of certain ingredients in the final volume of the parenteral nutrition admixture. Generally, pharmacy departments do not have the capability to routinely perform chemical analyses such as analyses of dextrose or electrolyte concentrations. Consequently, hospital or institutional laboratories may be called upon to perform these quality assurance tests. However, the methods in such laboratories are often designed for biological, not pharmaceutical, systems. Thus, their testing procedures shall be verified to meet the USP requirements stated in the individual monograph for the component being tested. For example, under Dextrose Injection, the following is stated: It contains not less than 95.0% and not more than 105.0% of the labeled amount of C6H12O6·H2O. The hospital or institutional chemistry laboratories must validate their methods to apply to this range and correct for their typical measurement of anhydrous dextrose versus dextrose monohydrate. Similar ranges and issues exist, for example, for injections of calcium gluconate, magnesium sulfate, and potassium chloride. The critical point is the use of USP references and possible laboratory procedural differences.
Precision
The intermediate precision of the ACD can be determined on the basis of the day-to-day variations in performance of the accuracy measures. Thus, compounding personnel shall keep a daily record of the above-described accuracy assessments and review the results over time. This review shall occur at least at weekly intervals to avoid potentially clinically significant cumulative errors over time. This is especially true for additives with a narrow therapeutic index, such as potassium chloride.
FINISHED PREPARATION RELEASE CHECKS AND TESTS
The following quality metrics shall be performed for all CSPs before they are dispensed or administered.
Inspection of Solution Dosage Forms and Review of Compounding Procedures
All CSPs that are intended to be solutions shall be visually examined for the presence of particulate matter and not administered or dispensed when such matter is observed. The prescription orders, written compounding procedure, preparation records, and expended materials used to make CSPs at all contamination risk levels are inspected for accuracy of correct identities and amounts of ingredients, aseptic mixing and sterilization, packaging, labeling, and expected physical appearance before they are administered or dispensed.
physical inspection
Finished CSPs are individually inspected in accordance with written procedures after compounding. If not distributed promptly, these CSPs are individually inspected just prior to leaving the storage area. Those CSPs that are not immediately distributed are stored in an appropriate location as described in the written procedures. Immediately after compounding, and as a condition of release, each CSP unit, where possible, should be inspected against lighted white or black background or both for evidence of visible particulates or other foreign matter. Prerelease inspection also includes container–closure integrity and any other apparent visual defect. CSPs with observed defects should be immediately discarded or marked and segregated from acceptable products in a manner that prevents their administration. When CSPs are not distributed promptly after preparation, a predistribution inspection is conducted to ensure that a CSP with defects, such as precipitation, cloudiness, and leakage, which may develop between the time of release and the time of distribution, is not released.
Compounding Accuracy Checks
Written procedures for double-checking compounding accuracy shall be followed for every CSP during preparation and immediately prior to release. The double-check system should meet state regulations and include label accuracy and accuracy of the addition of all drug products or ingredients used to prepare the finished product and their volumes or quantities. The used additive containers and, for those additives for which the entire container was not expended, the syringes used to measure the additive should be quarantined with the final products until the final product check is completed. Compounding personnel shall visually confirm that ingredients measured in syringes match the written order being compounded. Preferably, a person other than the compounder can verify that correct volumes of correct ingredients were measured to make each CSP. For example, compounding personnel would pull the syringe plunger back to the volume measured.
When practical, the accuracy of measurements is confirmed by weighing a volume of the measured fluid, then calculating that volume by dividing the weight by the accurate value of the density, or specific gravity, of the measured fluid. Correct density or specific gravity values programmed in ACDs, which measure by weight using the quotient of the programmed volume divided by the density or specific gravity, shall be confirmed to be accurate before and after delivering volumes of the liquids assigned to each channel or port. These volume accuracy checks and the following additional safety and accuracy checks in this section shall be included in the SOP manual of the CSP facility.
Sterility Testing
All high-risk level CSPs that are prepared in groups of more than 25 identical individual single-dose packages (e.g., ampuls, bags, syringes, vials) or in multiple-dose vials (MDVs) for administration to multiple patients or that are exposed longer than 12 hours at 2 to 8 and longer than 6 hours at warmer than 8 before they are sterilized shall meet the sterility test (see Sterility Tests 71) before they are dispensed or administered. The Membrane Filtration method is the method of choice where feasible (e.g., components are compatible with the membrane). A method not described in the USP may be used if verification results demonstrate that the alternative is at least as effective and reliable as the USP Membrane Filtration method or the USP Direct Inoculation of the Culture Medium method where the Membrane Filtration method is not feasible.
When high-risk level CSPs are dispensed before receiving the results of their sterility tests, there shall be a written procedure requiring daily observation of the incubating test specimens and immediate recall of the dispensed CSPs when there is any evidence of microbial growth in the test specimens. In addition, the patient and the physician of the patient to whom a potentially contaminated CSP was administered are notified of the potential risk. Positive sterility test results should prompt a rapid and systematic investigation of aseptic technique, environmental control, and other sterility assurance controls to identify sources of contamination and correct problems in the methods or processes.
Bacterial Endotoxin (Pyrogen) Testing
All high-risk level CSPs, except those for inhalation and ophthalmic administration, that are prepared in groups of more than 25 identical individual single-dose packages (e.g., ampuls, bags, syringes, vials) or in MDVs for administration to multiple patients or that are exposed longer than 12 hours at 2 to 8 and longer than 6 hours at warmer than 8 before they are sterilized shall be tested to ensure that they do not contain excessive bacterial endotoxins (see Bacterial Endotoxins Test 85 and Pyrogen Test 151). In the absence of a bacterial endotoxins limit in the official monograph or other CSP formula source, the CSP shall not exceed the amount of USP Endotoxin Units (per hour per kilogram of body weight or square meters of body surface area) specified in Bacterial Endotoxins Test 85 referenced above for the appropriate route of administration.
Identity and Strength Verification of Ingredients
Compounding facilities shall have at least the following written procedures for verifying the correct identity and quality of CSPs before they are dispensed and administered:
- That labels of CSPs bear correct names and amounts or concentrations of ingredients, the total volume, the BUD, the appropriate route(s) of administration, the storage conditions, and other information for safe use.
- That there are correct identities, purities, and amounts of ingredients by comparing the original written order with the written compounding record for the CSP.
- That correct fill volumes in CSPs and correct quantities of filled units of the CSPs were obtained. When the strength of finished CSPs cannot be confirmed to be accurate, based on the above three inspections, the CSPs shall be assayed by methods that are specific for the active ingredients.
STORAGE AND BEYOND-USE DATING
BUDs for compounded preparations are usually assigned on the basis of professional experience, which should include careful interpretation of appropriate information sources for the same or similar formulations (see Stability Criteria and Beyond-Use Dating under Pharmaceutical Compounding—Nonsterile Preparations 795). BUDs for CSPs are rarely based on preparation-specific chemical assay results, which are used with the Arrhenius equation to determine expiration dates (see General Notices and Requirements) for manufactured products. The majority of CSPs are aqueous solutions in which hydrolysis of dissolved ingredients is the most common chemical degradation reaction. The extent of hydrolysis and other heat-catalyzed degradation reactions at any particular time point in the life of a CSP represents the thermodynamic sum of exposure temperatures and durations. Such lifetime stability exposure is represented in the mean kinetic temperature calculation (see Pharmaceutical Calculations in Prescription Compounding 1160). Drug hydrolysis rates increase exponentially with arithmetic temperature increase; thus, exposure of a beta-lactam antibiotic solution for 1 day at controlled room temperature (see General Notices and Requirements) will have an equivalent effect on the extent of hydrolysis of approximately 3 to 5 days in cold temperatures (see General Notices and Requirements).
Personnel who prepare, dispense, and administer CSPs shall store them strictly in accordance with the conditions stated on the label of ingredient products and finished CSPs. When CSPs are known to have been exposed to temperatures warmer than the warmest labeled limit or to temperatures exceeding 40 (see General Notices and Requirements) for more than 4 hours, such CSPs should be discarded unless direct assay data or appropriate documentation confirms their continued stability.
Determining Beyond-Use Dates
BUDs and expiration dates are not the same (see General Notices and Requirements). Expiration dates for the chemical and physical stability of manufactured sterile products are determined from results of rigorous analytical and performance testing, and they are specific for a particular formulation in its container and at stated exposure conditions of illumination and temperature. When CSPs deviate from conditions in the approved labeling of manufactured products contained in CSPs, compounding personnel may consult the manufacturer of particular products for advice on assigning BUDs based on chemical and physical stability parameters. BUDs for CSPs that are prepared strictly in accordance with manufacturers' product labeling shall be those specified in that labeling or from appropriate literature sources or direct testing. BUDs for CSPs that lack justification from either appropriate literature sources or by direct testing evidence shall be assigned as described in Stability Criteria and Beyond-Use Dating under Pharmaceutical Compounding—Nonsterile Preparations 795.
In addition, compounding personnel may refer to applicable publications to obtain relevant stability, compatibility, and degradation information regarding the drug or its congeners. When assigning a beyond-use date, compounding personnel should consult and apply drug-specific and general stability documentation and literature where available, and they should consider the nature of the drug and its degradation mechanism, the container in which it is packaged, the expected storage conditions, and the intended duration of therapy (see Expiration Date and Beyond-Use Date under Labeling in the General Notices and Requirements). Stability information must be carefully interpreted in relation to the actual compounded formulation and conditions for storage and use. Predictions based on other evidence, such as publications, charts, and tables, would result in theoretical BUDs. Theoretically predicted beyond-use dating introduces varying degrees of assumptions and, hence, a likelihood of error or at least inaccuracy. The degree of error or inaccuracy would be dependent on the extent of differences between the CSPs' characteristics (e.g., composition, concentration of ingredients, fill volume, container type and material) and the characteristics of the products from which stability data or information is to be extrapolated. The greater the doubt of the accuracy of theoretically predicted beyond-use dating, the greater the need to determine dating periods experimentally. Theoretically predicted beyond-use dating periods should be carefully considered for CSPs prepared from nonsterile bulk active ingredients having therapeutic activity, especially where these CSPs are expected to be compounded routinely. When CSPs will be distributed to and administered in residential locations other than healthcare facilities, the effect of potentially uncontrolled and unmonitored temperature conditions shall be considered when assigning BUDs. It must be ascertained that CSPs will not be exposed to warm temperatures (see General Notices and Requirements) unless the compounding facility has evidence to justify stability of CSPs during such exposure.
It should be recognized that the truly valid evidence of stability for predicting beyond-use dating can be obtained only through product-specific experimental studies. Semiquantitative procedures such as thin-layer chromatography (TLC) may be acceptable for many CSPs. However, quantitative stability-indicating assays such as high-performance liquid chromatographic (HPLC) assays would be more appropriate for certain CSPs. Examples include CSPs with a narrow therapeutic index, where close monitoring or dose titration is required to ensure therapeutic effectiveness and to avoid toxicity; where a theoretically established beyond-use dating period is supported by only marginal evidence; or where a significant margin of safety cannot be verified for the proposed beyond-use dating period. In short, because beyond-use dating periods established from product-specific data acquired from the appropriate instrumental analyses are clearly more reliable than those predicted theoretically, the former approach is strongly urged to support dating periods exceeding 30 days.
To ensure consistent practices in determining and assigning BUDs, the compounding facility should have written policies and procedures governing the determination of the BUDs for all compounded products. When attempting to predict a theoretical BUD, a compounded or an admixed preparation should be considered as a unique system that has physical and chemical properties and stability characteristics that differ from its components. For example, antioxidant, buffering, or antimicrobial properties of a sterile vial for injection (SVI) might be lost upon its dilution, with the potential of seriously compromising the chemical stability of the SVI's active ingredient or the physical or microbiological stability of the SVI formulation in general. Thus, the properties stabilized in the SVI formulation usually cannot be expected to be carried over to the compounded or admixed preparation. Preparation-specific, experimentally determined stability data evaluation protocols are preferable to published stability information. Compounding personnel should consult general information chapter Pharmaceutical Stability 1150 for the appropriate stability parameters to be considered when initiating or evaluating a preparation-specific stability study.
Compounding personnel who assign BUDs to CSPs when lacking direct chemical assay results must critically interpret and evaluate the most appropriate available information sources to determine a conservative and safe BUD. The SOP manual of the compounding facility and each specific CSP formula record shall describe the general basis used to assign the BUD and storage conditions.
When manufactured MDVs (see Multiple-Dose Container under Preservation, Packaging, Storage, and Labeling in the General Notices and Requirements) of sterile ingredients are used in CSPs, the stoppers of the MDVs are inspected for physical integrity and disinfected by wiping with a sterile 70% IPA swab before each penetration with a sterile withdrawal device. When contaminants or abnormal properties are suspected or observed in MDVs, such MDVs shall be discarded. The BUD after initially entering or opening (e.g., needle puncturing) multiple-dose containers is 28 days (see Antimicrobial Effectiveness Testing 51) unless otherwise specified by the manufacturer.
Proprietary Bag and Vial Systems
The sterility storage and stability beyond-use times for attached and activated (where activated is defined as allowing contact of the previously separate diluent and drug contents) container pairs of drug products for intravascular administration (e.g., ADD-Vantage®, Mini Bag Plus®) shall be applied as indicated by the manufacturer. In other words, follow manufacturers' instructions for handling and storing ADD-Vantage®, Mini Bag Plus®, Add A Vial®, Add-Ease® products, and any others.
Monitoring Controlled Storage Areas
To ensure that product potency is retained through the manufacturer's labeled expiration date, compounding personnel shall monitor the drug storage areas within the compounding facility. Controlled temperature areas in compounding facilities include controlled room temperature, 20 to 25 with mean kinetic temperature 25; controlled cold temperature, 2 to 8 with mean kinetic temperature 8; cold temperature, 2 to 8; freezing temperature, 25 and 10 (see General Notices and Requirements) if needed to achieve freezing, and the media-specific temperature range for microbial culture media. A controlled temperature area shall be monitored at least once daily and the results documented on a temperature log. Additionally, compounding personnel shall note the storage temperature when placing the product into or removing the product from the storage unit in order to monitor any temperature aberrations. Suitable temperature recording devices may include a calibrated continuous recording device or a National Institute of Standards and Technology (NIST) calibrated thermometer that has adequate accuracy and sensitivity for the intended purpose, and it shall be properly calibrated at suitable intervals. If the compounding facility uses a continuous temperature recording device, compounding personnel shall verify at least once daily that the recording device itself is functioning properly.
The temperature-sensing mechanisms shall be suitably placed in the controlled temperature storage space to reflect accurately its true temperature. In addition, the compounding facility shall adhere to appropriate procedures of all controlled storage spaces to ensure that such spaces are not subject to significantly prolonged temperature fluctuations as may occur, for example, by leaving a refrigerator door open too long.
MAINTAINING STERILITY, PURITY, AND STABILITY OF DISPENSED AND DISTRIBUTED CSPs
This section summarizes the responsibilities of compounding facilities for maintaining quality and control of CSPs that are dispensed and administered within their parent healthcare organizations.
Compounding personnel shall ensure proper storage and security of CSPs prepared by or dispensed from the compounding facility until either their BUDs are reached or they are administered to patients. In fulfilling this general responsibility, the compounding facility is responsible for the proper packaging, handling, transport, and storage of CSPs prepared by or dispensed from it, including the appropriate education, training, and supervision of compounding personnel assigned to these functions. The compounding facility should assist in the education and training of noncompounding personnel responsible for carrying out any aspect of these functions.
Establishing, maintaining, and ensuring compliance with comprehensive written policies and procedures encompassing these responsibilities is a further responsibility of the compounding facility. Where noncompounding personnel are assigned tasks involving any of these responsibilities, the policies and procedures encompassing those tasks should be developed by compounding supervisors. The quality and control activities related to distribution of CSPs are summarized in the following five subsections. Activities or concerns that should be addressed as the compounding facility fulfills these responsibilities are as follows.
Packaging, Handling, and Transport
Inappropriate processes or techniques involved with packaging, handling, and transport can adversely affect quality and package integrity of CSPs. Although compounding personnel routinely perform many of the tasks associated with these functions, some tasks, such as transport, handling, and placement into storage, may be fulfilled by noncompounding personnel who are not under the direct administrative control of the compounding facility. Under these circumstances, appropriate SOPs shall be established by the compounding facility with the involvement of other departments or services whose personnel are responsible for carrying out those CSP-related functions for which the compounding facility has a direct interest. The performance of the noncompounding personnel is monitored for compliance to established policies and procedures.
The critical requirements that are unique to CSPs and that are necessary to ensure CSP quality and packaging integrity shall be addressed in SOPs. For example, techniques should be specified to prevent the depression of syringe plungers or dislodging of syringe tips during handling and transport. Additionally, disconnection of system components (e.g., where CSPs are dispensed with administration sets attached to them) shall be prevented through the BUD of the CSP. Foam padding or inserts are particularly useful where CSPs are transported by pneumatic tube systems. Regardless of the methods used, the compounding facility must evaluate their effectiveness and the reliability of the intended protection. Evaluation should be continuous—for example, through a surveillance system, including a system of problem reporting to the compounding facility.
Inappropriate transport and handling can adversely affect the quality of certain CSPs having unique stability concerns. For example, the physical shaking that might occur during pneumatic tube transport or undue exposure to heat or light must be addressed on a preparation-specific basis. Alternative transport modes or special packaging measures might be needed for the proper assurance of quality of these CSPs. The use of tamper-evident closures and seals on CSP ports can add an additional measure of security to ensure product integrity regardless of the transport method used.
Chemotoxic and other hazardous CSPs require safeguards to maintain the integrity of the CSP and to minimize the exposure potential of these products to the environment and to personnel who may come in contact with them. Transportation by pneumatic tube should be discouraged because of potential breakage and contamination. Special requirements associated with the packaging, transport, and handling of these agents include the prevention of accidental exposures or spills and the training of personnel in the event of an exposure or spill. Examples of special requirements of these agents also include exposure-reducing strategies such as the use of Luer lock syringes and connections, syringe caps, the capping of container ports, sealed plastic bags, impact-resistant containers, and cautionary labeling.
Use and Storage
The compounding facility is responsible for ensuring that CSPs in the patient-care setting maintain their quality until administered. The immediate labeling of the CSP container will display prominently and understandably the requirements for proper storage and expiration dating. Delivery and patient-care-setting personnel shall be properly trained to deliver the CSP to the appropriate storage location. Outdated and unused CSPs shall be returned to the compounding facility for disposition.
SOPs must exist to ensure that storage conditions in the patient-care setting are suitable for the CSP-specific storage requirements. Procedures include daily monitoring and documentation of drug storage refrigerators to ensure temperatures between 2 and 8 and the monthly inspection of all drug storage locations by compounding personnel. Inspections shall confirm compliance with appropriate storage conditions, separation of drugs and food, proper use of MDVs, and the avoidance of using single-dose products as MDVs. CSPs, as well as all other drug products, shall be stored in the patient-care area in such a way as to secure them from unauthorized personnel, visitors, and patients.
Readying for Administration
Procedures essential for generally ensuring quality, especially sterility assurance, when readying a CSP for its subsequent administration include proper hand washing, aseptic technique, site care, and change of administration sets. Additional procedures may also be essential for certain CSPs, devices, or techniques. Examples where such special procedures are needed include in-line filtration, the operation of automated infusion control devices, and the replenishment of CSPs into the reservoirs of implantable or portable infusion pumps. When CSPs are likely to be exposed to warmer than 30 for more than 1 hour during their administration to patients, the maintenance of their sterility and stability should be confirmed from either relevant and reliable sources or direct testing.
Redispensed CSPs
The compounding facility shall have the sole authority to determine when unopened, returned CSPs may be redispensed. Returned CSPs may be redispensed only when personnel responsible for sterile compounding can ensure that such CSPs are sterile, pure, and stable (contain labeled strength of ingredients). The following may provide such assurance: the CSPs were maintained under continuous refrigeration and protected from light, if required, and no evidence of tampering or any readying for use outside the compounding facility exists. Assignment of new storage times and BUDs that exceed the original dates for returned CSPs is permitted only when there is supporting evidence from sterility testing and quantitative assay of ingredients. Thus, initial preparation and thaw times should be documented and reliable measures should have been taken to prevent and detect tampering. Compliance with all procedures associated with maintaining product quality is essential. The CSPs shall not be redispensed if there is not adequate assurance that preparation quality and packaging integrity (including the connections of devices, where applicable) were continuously maintained between the time the CSPs left and the time they were returned. Additionally, CSPs shall not be redispensed if redispensing cannot be supported by the originally assigned BUD.
Education and Training
The assurance of CSPs' quality and packaging integrity is highly dependent on the proper adherence of all personnel to the pertinent SOPs. Compounding personnel shall design, implement, and maintain a formal education, training, and competency assessment program that encompasses all the functions and tasks addressed in the foregoing sections and all personnel to whom such functions and tasks are assigned. This program includes the assessment and documentation of procedural breaches, administration mishaps, side effects, allergic reactions, and complications associated with dosage or administration, such as extravasation. This program should be coordinated with the institution's adverse-events and incident reporting programs.
Packing and Transporting CSPs
The following sections describe how to maintain sterility and stability of CSPs until they are delivered to patient care locations for administration.
packing cpss for transit
When CSPs are distributed to locations outside the premises in which they are compounded, compounding personnel select packing containers and materials that are expected to maintain physical integrity, sterility, and stability of CSPs during transit. Packing is selected that simultaneously protects CSPs from damage, leakage, contamination, and degradation, and protects personnel who transport packed CSPs from harm. The SOP manual of the compounding facility specifically describes appropriate packing containers and insulating and stuffing materials, based on information from product specifications, vendors, and experience of compounding personnel. Written instructions that clearly explain how to safely open containers of packed CSPs are provided to patients and other recipients.
transit of cpss
Compounding facilities that ship CSPs to locations outside their own premises shall select modes of transport that are expected to deliver properly packed CSPs in undamaged, sterile, and stable condition to recipients.
Compounding personnel should ascertain that temperatures of CSPs during transit by the selected mode will not exceed the warmest temperature specified on the storage temperature range on CSP labels. It is recommended that compounding personnel communicate directly with the couriers to learn shipping durations and exposure conditions that CSPs may encounter.
Compounding personnel shall include specific handling and exposure instructions on the exteriors of containers packed with CSPs to be transported and obtain reasonable assurance of compliance therewith from transporters. Compounding personnel shall periodically review the delivery performance of couriers to ascertain that CSPs are being efficiently and properly transported.
Storage in Locations Outside Compounding Facilities
Compounding facilities that ship CSPs to patients and other recipients outside their own premises shall ascertain or provide, whichever is appropriate, the following assurances:
- Labels and accessory labeling for CSPs include clearly readable BUDs, storage instructions, and disposal instructions for out-of-date units.
- Each patient or other recipient is able to store the CSPs properly, including the use of a properly functioning refrigerator and freezer if CSPs are labeled for such storage.
PATIENT OR CAREGIVER TRAINING
A formal training program is provided as a means to ensure understanding and compliance with the many special and complex responsibilities placed on the patient or caregiver for the storage, handling, and administration of CSPs. The instructional objectives for the training program include all home care responsibilities expected of the patient or caregiver and is specified in terms of patient or caregiver competencies.
Upon the conclusion of the training program, the patient or caregiver should, correctly and consistently, be able to do the following:
- Describe the therapy involved, including the disease or condition for which the CSPs are prescribed, goals of therapy, expected therapeutic outcome, and potential side effects of the CSPs.
- Inspect all drug products, CSPs, devices, equipment, and supplies on receipt to ensure that proper temperatures were maintained during transport and that goods received show no evidence of deterioration or defects.
- Handle, store, and monitor all drug products, CSPs, and related supplies and equipment in the home, including all special requirements related to same.
- Visually inspect all drug products, CSPs, devices, and other items the patient or caregiver is required to use immediately prior to administration in a manner to ensure that all items are acceptable for use. For example, CSPs must be free from leakage, container cracks, particulates, precipitate, haziness, discoloration, or other deviations from the normal expected appearance, and the immediate packages of sterile devices must be completely sealed, with no evidence of loss of package integrity.
- Check labels immediately prior to administration to ensure the right drug, dose, patient, and time of administration.
- Clean the in-home preparation area, scrub hands, use proper aseptic technique, and manipulate all containers, equipment, apparatus, devices, and supplies used in conjunction with administration.
- Employ all techniques and precautions associated with CSP administration; for example, preparing supplies and equipment, handling of devices, priming the tubing, and discontinuing an infusion.
- Care for catheters, change dressings, and maintain site patency as indicated.
- Monitor for and detect occurrences of therapeutic complications such as infection, phlebitis, electrolyte imbalance, and catheter misplacement.
- Respond immediately to emergency or critical situations such as catheter breakage or displacement, tubing disconnection, clot formation, flow blockage, and equipment malfunction.
- Know when to seek and how to obtain professional emergency services or professional advice.
- Handle, contain, and dispose of wastes, such as needles, syringes, devices, biohazardous spills or residuals, and infectious substances.
Training programs include a hands-on demonstration and practice with actual items that the patient or caregiver is expected to use, such as CSP containers, devices, and equipment. The patient or caregiver practices aseptic and injection technique under the direct observation of a health professional.
The compounding facility, in conjunction with nursing or medical personnel, is responsible for ensuring initially and on an ongoing basis that the patient or caregiver understands, has mastered, and is capable of and willing to comply with all of these home care responsibilities. This is achieved through a formal, written assessment program. All specified competencies in the patient or caregiver training program are formally assessed. The patient or caregiver is expected to demonstrate to appropriate healthcare personnel mastery of assigned activities before being allowed to administer CSPs unsupervised by a health professional.
Printed material such as checklists or instructions provided during training may serve as continuing post-training reinforcement of learning or as reminders of specific patient or caregiver responsibilities. Post-training verbal counseling can also be used periodically, as appropriate, to reinforce training and to ensure continuing correct and complete fulfillment of responsibilities.
PATIENT MONITORING AND ADVERSE EVENTS REPORTING
Compounding facilities shall clinically monitor patients treated with CSPs according to the regulations and guidelines of their respective state healthcare practitioner licensure boards or of accepted standards of practice. Compounding facilities shall provide patients and other recipients of CSPs with a way to address their questions and report any concerns that they may have with CSPs and their administration devices.
The SOP manuals of compounding facilities shall describe specific instructions for receiving, acknowledging, and dating receipts, and for recording, or filing, and evaluating reports of adverse events and of the quality of preparation claimed to be associated with CSPs. Reports of adverse events with CSPs shall be reviewed promptly and thoroughly by compounding supervisors to correct and prevent future occurrences. Compounding personnel are encouraged to participate in adverse event reporting and product defects programs of the FDA and USP.
QUALITY ASSURANCE (QA) PROGRAM
A provider of CSPs shall have in place a formal QA program intended to provide a mechanism for monitoring, evaluating, correcting, and improving the activities and processes described in this chapter. Emphasis in the QA program is placed on maintaining and improving the quality of systems and the provision of patient care. In addition, the QA program ensures that any plan aimed at correcting identified problems also includes appropriate follow-up to make certain that effective corrective actions were performed.13
Characteristics of a QA program include the following:
- Formalization in writing;
- Consideration of all aspects of the preparations and dispensing of products as described in this chapter, including environmental testing and verification results;
- Description of specific monitoring and evaluation activities;
- Specification of how results are to be reported and evaluated;
- Identification of appropriate follow-up mechanisms when action limits or thresholds are exceeded; and
- Delineation of the individuals responsible for each aspect of the QA program.
In developing a specific plan, focus is on establishing objective, measurable indicators for monitoring activities and processes that are deemed high risk, high volume, or problem prone. In general, the selection of indicators and the effectiveness of the overall QA program is reassessed on an annual basis.
ABBREVIATIONS AND ACRONYMS
| ACD | automated compounding device |
| ACPH | air changes per hour |
| ALARA | as low as reasonably achievable |
| ASHRAE | American Society of Heating, Refrigerating and Air-Conditioning Engineers |
| BI | biological indicator |
| BSC | biological safety cabinet |
| BUD | beyond-use date |
| CACI | compounding aseptic containment isolator |
| CAI | compounding aseptic isolator |
| CDC | Centers for Disease Control and Prevention |
| CETA | Controlled Environment Testing Association |
| cfu | colony-forming unit(s) |
| CSP | compounded sterile preparation |
| CSTD | closed-system vial-transfer device |
| DCA | direct compounding area |
| ECV | endotoxin challenge vial |
| EU | Endotoxin Unit |
| FDA | Food and Drug Administration |
| HEPA | high efficiency particulate air |
| HICPAC | Healthcare Infection Control Practices Advisory Committee |
| HVAC | heating, ventilation, and air conditioning |
| IPA | isopropyl alcohol |
| ISO | International Organization for Standardization |
| LAFW | laminar airflow workbench |
| MDVs | multiple-dose vials |
| MMWR | Morbidity and Mortality Weekly Report |
| NIOSH | National Institute for Occupational Safety and Health |
| NIST | National Institute of Standards and Technology |
| PEC | primary engineering control |
| PET | positron emission tomography |
| PPE | personnel protective equipment |
| psi | pounds per square inch |
| QA | quality assurance |
| SOP | standard operating procedure |
| SVI | sterile vial for injection |
| TSA | trypticase soy agar |
| USP | United States Pharmacopeia |
APPENDICES
Appendix I. Principal Competencies, Conditions, Practices, and Quality Assurances That Are Required († “shall”) and Recommended (‡ “should”) in USP Chapter797
Appendix I. Principal Competencies, Conditions, Practices, and Quality Assurances That Are Required († “shall”) and Recommended (‡ “should”) in USP Chapter
| note—This tabular appendix selectively abstracts and condenses the full text of |
| INTRODUCTION |
| ‡ Chapter purpose is to prevent harm and death to patients treated with CSPs. |
| † Chapter pertains to preparation, storage, and transportation, but not administration, of CSPs. |
| † Personnel and facilities to which |
| † Types of preparations designated to be CSPs according to their physical forms, and their sites and routes of administration to patients. |
| † Compounding personnel must be meticulously conscientious to preclude contact contamination of CSPs both within and outside ISO Class 5 areas. |
| ORGANIZATION |
| † All compounding personnel shall be responsible for understanding fundamental practices and precautions within USP |
| DEFINITIONS |
| † Twenty-eight terms are defined and integral to complying with USP |
| RESPONSIBILITY OF COMPOUNDING PERSONNEL |
| † Practices and quality assurances required to prepare, store, and transport CSPs that are sterile, and acceptably accurate, pure, and stable. |
| CSP MICROBIAL CONTAMINATION RISK LEVELS |
| † Proper training and evaluation of personnel, proper cleansing and garbing of personnel, proper cleaning and disinfecting of compounding work environments, and proper maintenance and monitoring of controlled environmental locations (all of which are detailed in their respective sections). |
| Low-Risk Level CSPs |
| † Aseptic manipulations within an ISO Class 5 environment using three or fewer sterile products and entries into any container. |
| † In absence of passing sterility test, store not more than 48 hours at controlled room temperature, 14 days at cold temperature, and 45 days in solid frozen state at |
| † Media-fill test at least annually by compounding personnel. |
| Low-Risk Level CSPs with 12-Hour or Less BUD |
| † Fully comply with all four specific criteria. |
| ‡ Sinks should not be located adjacent to the ISO Class 5 primary engineering control. |
| ‡ Sinks should be separated from the immediate area of the ISO Class 5 primary engineering control device. |
| Medium-Risk Level CSPs |
| † Aseptic manipulations within an ISO Class 5 environment using prolonged and complex mixing and transfer, more than three sterile products and entries into any container, and pooling ingredients from multiple sterile products to prepare multiple CSPs. |
| † In absence of passing sterility test, store not more than 30 hours at controlled room temperature, 9 days at cold temperature, and 45 days in solid frozen state at |
| †Media-fill test at least annually by compounding personnel. |
| High-Risk Level CSPs |
| † Confirmed presence of nonsterile ingredients and devices, or confirmed or suspected exposure of sterile ingredients for more than one hour to air quality inferior to ISO Class 5 before final sterilization. |
| † Sterilization method verified to achieve sterility for the quantity and type of containers. |
| † Meet allowable limits for bacterial endotoxins. |
| † Maintain acceptable strength and purity of ingredients and integrity of containers after sterilization. |
| † In absence of passing sterility test, store not more than 24 hours at controlled room temperature, 3 days at cold temperature, and 45 days in solid frozen state at |
| † Media-fill test at least semiannually by compounding personnel. |
| PERSONNEL TRAINING AND EVALUATION IN ASEPTIC MANIPULATIONS SKILLS |
| † Pass didactic, practical skill assessment and media-fill testing initially, followed by an annual assessment for a low- and medium-risk level compounding and semi-annual assessment for high-risk level compounding. |
| † Compounding personnel who fail written tests, or whose media-fill test vials result in gross microbial colonization, shall be immediately reinstructed and re-evaluated by expert compounding personnel to ensure correction of all aseptic practice deficiencies. |
| IMMEDIATE-USE CSPs |
| † Fully comply with all six specified criteria. |
| SINGLE-DOSE AND MULTIPLE-DOSE CONTAINERS |
| † Beyond-use date 28 days, unless specified otherwise by the manufacturer, for closure sealed multiple-dose containers after initial opening or entry. |
| † Beyond-use time of 6 hours, unless specified otherwise by the manufacturer, for closure sealed single-dose containers in ISO Class 5 or cleaner air after initial opening or entry. |
| † Beyond-use time of 1 hour for closure sealed single-dose containers after being opened or entered in worse than ISO Class 5 air. |
| † Storage of opened single-dose ampuls is not permitted. |
| HAZARDOUS DRUGS AS CSPs |
| † Appropriate personnel protective equipment. |
| † Appropriate primary engineering controls (BSCs and CACIs) are used for concurrent personnel protection and exposure of critical sites. |
| † Hazardous drugs shall be stored separately from other inventory in a manner to prevent contamination and personnel exposure. |
| † At least 0.01 inch water column negative pressure and 12 air changes per hour in non-cleanrooms in which CACIs are located. |
| † Hazardous drugs shall be handled with caution at all times using appropriate chemotherapy gloves during receiving, distribution, stocking, inventorying, preparing for administration, and disposal. |
| † Hazardous drugs shall be prepared in an ISO Class 5 environment with protective engineering controls in place, and following aseptic practices specified for the appropriate contamination risk levels. |
| † Access to drug preparation areas shall be limited to authorized personnel. |
| † A pressure indicator shall be installed that can readily monitor room pressurization, which is documented daily. |
| † Annual documentation of full training of personnel regarding storage, handling, and disposal of hazardous drugs. |
| † When used, a CSTD shall be used in an ISO Class 5 primary engineering control device. |
| † At least 0.01 inch water column negative pressure is required for compounding of hazardous drugs. |
| ‡ Negative-pressure buffer area is not required for low-volume compounding operations when CSTD is used in BSC or CACI. |
| † Compounding personnel of reproductive capability shall confirm in writing that they understand the risks of handling hazardous drugs. |
| † Disposal of all hazardous drug wastes shall comply with all applicable federal and state regulations. |
| ‡ Total external exhaust of primary engineering controls. |
| ‡ Assay of surface wipe samples every 6 months. |
| RADIOPHARMACEUTICALS AS CSPs |
| † Positron Emission Tomography is according to USP chapter |
| † Appropriate primary engineering controls and radioactivity containment and shielding. |
| † Radiopharmaceuticals compounded from sterile components, in closed sterile containers, with volume of 100 mL or less for a single-dose injection or not more than 30 mL taken from a multiple-dose container shall be designated as and conform to the standards for low-risk level CSPs. |
| † Radiopharmaceutical vials, designed for multi-use, compounded with technetium-99m, exposed to ISO Class 5 environment and punctured by needles with no direct contact contamination may be used up to the time indicated by manufacturers' recommendations. |
| † Location of primary engineering controls permitted in ISO Class 8 controlled environment. |
| † Technetium-99m/Molybdenum-99 generators used according to manufacturer, state, and federal requirements. |
| † Radiopharmaceuticals prepared as low-risk level CSPs with 12-hour or less BUD shall be prepared in a segregated compounding area. |
| † Materials and garb exposed in patient-care and treatment area shall not cross a line of demarcation into the segregated compounding area. |
| † Technetium-99m/Molybdenum-99 generators must be eluted in ISO Class 8 conditions. |
| † Segregated compounding area will be designated with a line of demarcation. |
| ‡ Storage and transport of properly shielded vials of radiopharmaceutical CSPs may occur in a limited access ambient environment without a specific ISO class designation. |
| ALLERGEN EXTRACTS AS CSPs |
| † Allergen extracts as CSPs are not subject to the personnel, environmental, and storage requirements for all CSP Microbial Contamination Risk Levels when certain criteria are met. |
| VERIFICATION OF COMPOUNDING ACCURACY AND STERILITY |
| † Review labels and document correct measurements, aseptic manipulations, and sterilization procedures to confirm correct identity, purity, and strength of ingredients in, and sterility of, CSPs. |
| ‡ Assay finished CSPs to confirm correct identity and, or, strength of ingredients. |
| ‡ Sterility test finished CSPs. |
| Sterilization Methods |
| † Verify that methods achieve sterility while maintaining appropriate strength, purity, quality, and packaging integrity. |
| ‡ Prove effectiveness by USP chapter |
| Sterilization of High-Risk Level CSPs by Filtration |
| † Nominal 0.2-µm pore size sterile membranes that are chemically and physically compatible with the CSP. |
| † Complete rapidly without filter replacement. |
| † Subject filter to manufacturer's recommended integrity test (e.g., bubble point test) after filtering CSPs. |
| Sterilization of High-Risk Level CSPs by Steam |
| † Test to verify the mass of containers to be sterilized will be sterile after the selected exposure duration in the particular autoclave. |
| † Ensure live steam contacts all ingredients and surfaces to be sterilized. |
| † Pass solutions through a 1.2-µm or smaller nominal pore size filter into final containers to remove particulates before sterilization. |
| † Heated filtered air shall be evenly distributed throughout the chamber by a blower device. |
| † Dry heat shall only be used for those materials that cannot be sterilized by steam, when the moisture would either damage or be impermeable to the materials. |
| † Sufficient space shall be left between materials to allow for good circulation of the hot air. |
| † The description of dry heat sterilization conditions and duration for specific CSPs shall be included in written documentation in the compounding facility. The effectiveness of dry heat sterilization shall be verified using appropriate biological indicators and other confirmation. |
| ‡ The oven should be equipped with a system for controlling temperature and exposure period. |
| Depyrogenation by Dry Heat |
| † Dry heat depyrogenation shall be used to render glassware or containers, such as vials free from pyrogens as well as viable microbes. |
| † The description of the dry heat depyrogenation cycle and duration for specific load items shall be included in written documentation in the compounding facility. |
| † The effectiveness of the dry heat depyrogenation cycle shall be verified using endotoxin challenge vials (ECVs). |
| ‡ The bacterial endotoxin test should be performed on the ECVs to verify the cycle is capable of achieving a 3 log reduction in endotoxin. |
| ENVIRONMENTAL QUALITY AND CONTROL |
| Exposure of Critical Sites |
| † ISO Class 5 or better air. |
| † Preclude direct contact (e.g., touch and secretions) contamination. |
| ISO Class 5 Air Sources, Buffer Areas, and Ante-Areas |
| † A buffer area is an area that provides at least ISO Class 7 air quality. |
| † New representations of facility layouts. |
| † Each compounding facility shall ensure that each source of ISO Class 5 environment for exposure of critical sites and sterilization by filtration is properly located, operated, maintained, monitored, and verified. |
| † Devices (e.g., computers and printers) and objects (e.g., carts and cabinets) can be placed in buffer areas and shall be verified by testing or monitoring. |
| Viable and Nonviable Environmental Sampling (ES) Testing |
| † Environmental sampling shall occur as part a comprehensive quality management program and shall occur minimally when several conditions exist. |
| ‡ The ES program should provide information to staff and leadership to demonstrate that the engineering controls are maintaining an environment within the compounding area that consistently maintains acceptably low viable and nonviable particle levels. |
| Environmental Nonviable Particle Testing Program |
| † Certification and testing of primary (LAFWs, BSCs, CAIs and CACIs) and secondary engineering controls (buffer and ante areas) shall be performed by a qualified individual no less than every six months and whenever the device or room is relocated, altered, or major service to the facility is performed. Certification procedures such as those outlined in the CETA Certification Guide for Sterile Compounding Facilities (CAG-003-2006) shall be used. |
| Total Particle Counts |
| † Certification that each ISO classified area (e.g., ISO Class 5, 7 and 8) is within established guidelines shall be performed no less than every 6 months and whenever the LAFW, BSC, CAI, or CACI is relocated or the physical structure of the buffer room or ante-area has been altered. |
| † Testing shall be performed by qualified operators using current, state-of-the-art electronic equipment with results meeting ISO Class 5, 7, or 8 depending on the requirements of the area. |
| † All certification records shall be maintained and reviewed by supervising personnel or other designated employee to ensure that the controlled environments comply with the proper air cleanliness, room pressures, and air changes per hour. |
| Pressure Differential Monitoring |
| † A pressure gauge or velocity meter shall be installed to monitor the pressure differential or airflow between the buffer area and ante-area, and the ante-area and the general environment outside the compounding area. |
| † The results shall be reviewed and documented on a log at least every work shift (minimum frequency shall be at least daily) or by a continuous recording device. |
| † The pressure between the ISO Class 7 and general pharmacy area shall not be less than 5 Pa (0.02 inch water column (w.c.)). |
| † In facilities where low- and medium-risk level CSPs are prepared, differential airflow shall maintain a minimum velocity of 0.2 meter/second (40 fpm) between buffer area and ante-area. |
| Environmental Viable Airborne Particle Testing Program—Sampling Plan |
| † An appropriate environmental sampling plan shall be developed for airborne viable particles based on a risk assessment of compounding activities performed. |
| † Selected sampling sites shall include locations within each ISO Class 5 environment and in the ISO Class 7 and 8 areas, and the segregated compounding areas at greatest risk of contamination (e.g., work areas near the ISO Class 5 environment, counters near doors, pass-through boxes). |
| † The plan shall include sample location, method of collection, frequency of sampling, volume of air sampled, and time of day as related to activity in the compounding area and action levels. |
| ‡ It is recommended that compounding personnel refer to USP Chapter Microbiological Evaluation of Clean Rooms and Other Controlled Environments |
| Growth Media |
| † A general microbiological growth medium such as Soybean–Casein Digest Medium (also known as trypticase soy broth (TSB) or agar (TSA)) shall be used to support the growth of bacteria. |
| † Malt extract agar (MEA) or some other media that supports the growth of fungi shall be used in high-risk level compounding environments. |
| † Media used for surface sampling shall be supplemented with additives to neutralize the effects of disinfecting agents (e.g., TSA with lecithin and polysorbate 80). |
| Viable Air Sampling |
| † Evaluation of airborne microorganisms using volumetric collection methods in the controlled air environments shall be performed by properly trained individuals for all compounding risk levels. |
| † Impaction shall be the preferred method of volumetric air sampling. |
| † For low-, medium-, and high-risk level compounding, air sampling shall be performed at locations that are prone to contamination during compounding activities and during other activities like staging, labeling, gowning, and cleaning. |
| † Locations shall include zones of air backwash turbulence within laminar airflow workbench and other areas where air backwash turbulence may enter the compounding area. |
| † For low-risk level CSPs with 12-hour or less BUD, air sampling shall be performed at locations inside the ISO Class 5 environment and other areas that are in close proximity to the ISO class 5 environment, during the certification of the primary engineering control. |
| ‡ Consideration should be given to the overall effect the chosen sampling method will have on the unidirectional airflow within a compounding environment. |
| Air Sampling Devices |
| † The instructions in the manufacturer's user manual for verification and use of electric air samplers that actively collect volumes of air for evaluation shall be followed. |
| † A sufficient volume of air (400–1000 liters) shall be tested at each location in order to maximize sensitivity. |
| ‡ It is recommended that compounding personnel also refer to USP Chapter |
| Air Sampling Frequency and Process |
| † Air sampling shall be performed at least semiannually (i.e. every 6 months), as part of the re-certification of facilities and equipment for area where primary engineering controls are located. |
| † A sufficient volume of air shall be sampled and the manufacturer's guidelines for use of the electronic air sampling equipment followed. |
| ‡ Any facility construction or equipment servicing may require the need to perform air sampling during these events. |
| Incubation Period |
| † The microbial growth media plates used to collect environmental sampling are recovered, covers secured (e.g., taped), inverted, and incubated at a temperature and for a time period conducive to multiplication of microorganisms. |
| † The number of discrete colonies of microorganisms shall be counted and reported as colony-forming units (cfu) and documented on an environmental monitoring form. Counts from air monitoring need to be transformed into cfu/cubic meter of air and evaluated for adverse trends. |
| ‡ TSA should be incubated at 35 |
| ‡ MEA or other suitable fungal media should be incubated at 28 |
| Action Levels, Documentation and Data Evaluation |
| † Sampling data shall be collected and reviewed on a periodic basis as a means of evaluating the overall control of the compounding environment. |
| † Competent microbiology personnel shall be consulted if an environmental sampling consistently shows elevated levels of microbial growth. |
| † An investigation into the source of the environmental contamination shall be conducted. |
| ‡ Any cfu count that exceeds its respective action level should prompt a re-evaluation of the adequacy of personnel work practices, cleaning procedures, operational procedures, and air filtration efficiency within the aseptic compounding location. |
| ‡ Table titled, Recommended Action Levels for Microbial Contamination should only be used as a guideline |
| Facility Design and Environmental Controls |
| † Compounding facilities are physically designed and environmentally controlled to minimize airborne contamination from contacting critical sites. |
| † Compounding facilities shall provide a comfortable and well-lighted working environment, which typically includes a temperature of 20 |
| † Primary engineering controls provide unidirectional (i.e., laminar) HEPA air at a velocity sufficient to prevent airborne particles from contacting critical sites. |
| † In situ air pattern analysis via smoke studies shall be conducted at the critical area to demonstrate unidirectional airflow and sweeping action over and away from the product under dynamic conditions. |
| † Policies and procedures for maintaining and working within the primary engineering control area shall be written and followed. The policies and procedures will be determined by the scope and risk levels of the aseptic compounding activities used during the preparation of the CSPs. |
| † The principles of HEPA-filtered unidirectional airflow in the work environment shall be understood and practiced in the compounding process in order to achieve the desired environmental conditions. |
| † Clean rooms for nonhazardous and nonradioactive CSPs are supplied with HEPA that enters from ceilings with return vents low on walls, and that provides not less than 30 air changes per hour. |
| † Buffer areas maintain 0.02- to 0.05-inch water column positive pressure, and do not contain sinks or drains. |
| † Air velocity from buffer rooms or zones to ante-areas is at least 40 feet/minute. |
| † The primary engineering controls shall be placed within a buffer area in such a manner as to avoid conditions that could adversely affect their operation. |
| † The primary engineering controls shall be placed out of the traffic flow and in a manner to avoid disruption from the HVAC system and room cross-drafts. |
| † HEPA-filtered supply air shall be introduced at the ceiling. |
| † All HEPA filters shall be efficiency tested using the most penetrating particle size and shall be leak tested at the factory and then leak tested again in situ after installation. |
| † Activities and tasks carried out within the buffer area shall be limited to only those necessary when working within a controlled environment. |
| † Only the furniture, equipment, supplies, and other material required for the compounding activities to be performed shall be brought into the room. |
| † Surfaces and essential furniture in buffer rooms or zones and clean rooms shall be nonporous, smooth, nonshedding, impermeable, cleanable, and resistant to disinfectants. |
| † The surfaces of ceilings, walls, floors, fixtures, shelving, counters, and cabinets in the buffer area shall be smooth, impervious, free from cracks and crevices, and nonshedding, thereby promoting cleanability, and minimizing spaces in which microorganisms and other contaminants may accumulate. |
| † The surfaces shall be resistant to damage by disinfectant agents. |
| † Junctures of ceilings to walls shall be coved or caulked to avoid cracks and crevices where dirt can accumulate. |
| † Ceiling tiles shall be caulked around each perimeter to seal them to the support frame. |
| † The exterior lens surface of ceiling lighting fixtures shall be smooth, mounted flush, and sealed. |
| † Any other penetrations through the ceiling or walls shall be sealed. |
| † The buffer area shall not contain sources of water (sinks) or floor drains. Work surfaces shall be constructed of smooth, impervious materials, such as stainless steel or molded plastic, so that they are easily cleaned and disinfected. |
| † Carts shall be of stainless steel wire, nonporous plastic, or sheet metal construction with good quality, cleanable casters to promote mobility. |
| † Storage shelving, counters, and cabinets shall be smooth, impervious, free from cracks and crevices, nonshedding, cleanable, and disinfectable. |
| † Their number, design, and manner of installation the itmes above shall promote effective cleaning and disinfection. |
| ‡ If ceilings consist of inlaid panels, the panels should be impregnated with a polymer to render them impervious and hydrophobic. |
| ‡ Dust-collecting overhangs, such as ceiling utility pipes, or ledges, such as windowsills, should be avoided. |
| ‡ Air returns should be mounted low on the wall creating a general top-down dilution of room air with HEPA-filtered make-up air. |
| Placement of Primary Engineering Controls Within ISO Class 7 Buffer Areas |
| † Primary engineering controls for nonhazardous and nonradioactive CSPs are located in buffer areas, except for CAIs that are proven to maintain ISO Class 5 air when particle counts are sampled 6 to 12 inches upstream of critical site exposure areas during performance of normal inward and outward transfer of materials, and compounding manipulations when such CAIs are located in air quality worse than ISO Class 7. |
| † Presterilization procedures for high-risk level CSPs, such as weighing and mixing, shall be completed in no worse than an ISO Class 8 environment. |
| † Primary engineering controls shall be located out of traffic patterns and away from room air currents that could disrupt the intended airflow patterns. |
| † When isolators are used for sterile compounding, the recovery time to achieve ISO Class 5 air quality shall be documented and internal procedures developed to ensure that adequate recovery time is allowed after material transfer before and during compounding operations. |
| † When compounding activities require the manipulation of a patient's blood-derived or other biological material (e.g., radiolabeling a patient's or a donor's white blood cells), the manipulations shall be clearly separated from routine material-handling procedures and equipment used in CSP preparation activities, and they shall be controlled by specific standard operating procedures in order to avoid any cross-contamination. |
| † Food, drinks, and items exposed in patient care areas, and unpacking of bulk supplies and personnel cleansing and garbing are prohibited from buffer areas or rooms. |
| † Demarcation designation between buffer areas or rooms and ante-areas. |
| † Antiseptic hand cleansing and sterile gloves in buffer areas or rooms. |
| ‡ Packaged compounding supplies and components, such as needles, syringes, tubing sets, and small- and large-volume parenterals, should be uncartoned and wiped down with a disinfectant that does not leave a residue (e.g., sterile 70% IPA) when possible in an ante-area, of ISO Class 8 air quality, before being passed into the buffer areas. |
| Cleaning and Disinfecting the Sterile Compounding Areas |
| † Trained personnel write detailed procedures including cleansers, disinfectants, and non-shedding wipe and mop materials. |
| † Cleaning and disinfecting surfaces in the LAFWs, BSCs, CAIs, and CACIs shall be cleaned and disinfected frequently, including at the beginning of each work shift, before each batch preparation is started, every 30 minutes during continuous compounding periods of individual CSPs, when there are spills, and when surface contamination is known or suspected from procedural breaches. |
| † Trained compounding personnel are responsible for developing, implementing, and practicing the procedures for cleaning and disinfecting the DCAs written in the SOPs. |
| † Cleaning and disinfecting shall occur before compounding is performed. Items shall be removed from all areas to be cleaned, and surfaces shall be cleaned by removing loose material and residue from spills, e.g., water-soluble solid residues are removed with Sterile Water (for Injection or Irrigation) and low-shedding wipes. This shall be followed by wiping with a residue-free disinfecting agent, such as sterile 70% IPA, which is allowed to dry before compounding begins. |
| † Work surfaces in ISO Class 7 and 8 areas and segregated compounding areas are cleaned at least daily. |
| † Dust and debris shall be removed when necessary from storage sites for compounding ingredients and supplies, using a method that does not degrade the ISO Class 7 or 8 air quality. |
| † Floors in ISO Class 7 and 8 areas are cleaned daily when no compounding occurs. |
| † IPA (70% isopropyl alcohol) remains on surfaces to be disinfected for at least 30 seconds before such surfaces are used to prepare CSPs. |
| † Emptied shelving, walls, and ceilings in ante-areas are cleaned and disinfected at least monthly. |
| † Mopping shall be performed by trained personnel using approved agents and procedures described in the written SOPs. |
| † Cleaning and disinfecting agents, their schedules of use and methods of application shall be in accordance with written SOPs and followed by custodial and/or compounding personnel. |
| † All cleaning materials, such as wipers, sponges, and mops, shall be nonshedding, preferably composed of synthetic micro fibers, and dedicated to use in the buffer area, or ante-area, and segregated compounding areas and shall not be removed from these areas except for disposal. |
| † If cleaning materials are reused (e.g., mops), procedures shall be developed (based on manufacturer recommendations) that ensure that the effectiveness of the cleaning device is maintained and repeated use does not add to the bioburden of the area being cleaned. |
| † Supplies and equipment removed from shipping cartons shall be wiped with a suitable disinfecting agent (e.g., sterile 70% IPA) delivered from a spray bottle or other suitable delivery method. |
| † After the disinfectant is sprayed or wiped on a surface to be disinfected, the disinfectant shall be allowed to dry, and during this time the item shall not be used for compounding purposes. |
| † Sterile 70% IPA wetted gauze pads or other particle-generating material shall not be used to disinfect the sterile entry points of packages and devices. |
| Personnel Cleansing and Garbing |
| † Personnel shall also be thoroughly competent and highly motivated to perform flawless aseptic manipulations with ingredients, devices, and components of CSPs. |
| † Personnel with rashes, sunburn, weeping sores, conjunctivitis, active respiratory infection, and cosmetics are prohibited from preparing CSPs. |
| † Compounding personnel shall remove personal outer garments; cosmetics; artificial nails; hand, wrist, and body jewelry that can interfere with the fit of gowns and gloves; and visible body piercing above the neck. |
| † Order of compounding garb and cleansing in ante-area: shoes or shoe covers, head and facial hair covers, face mask, fingernail cleansing, hand and forearm washing and drying; non-shedding gown. |
| † Order of cleansing and gloving in buffer room or area: hand cleansing with a persistently active alcohol-based product with persistent activity; allow hands to dry; don sterile gloves. |
| † Routinely disinfect gloves with sterile 70% IPA after contacting nonsterile objects. |
| † Inspect gloves for holes and replace when breaches are detected. |
| † Personnel repeat proper procedures after they are exposed to direct contact contamination or worse than ISO Class 8 air. |
| † These requirements are exempted only for immediate-use CSPs and CAIs for which manufacturers provide written documentation based on validated testing that such personnel practices are not required to maintain sterility in CSPs. |
| Personnel Training and Competency Evaluation of Garbing, Aseptic Work Practices and Cleaning/Disinfection Procedures |
| † Personnel who prepare CSPs shall be trained conscientiously and skillfully by expert personnel, multi-media instructional sources, and professional publications in the theoretical principles and practical skills of garbing procedures, aseptic work practices, achieving and maintaining ISO Class 5 environmental conditions, and cleaning and disinfection procedures. |
| † This training shall be completed and documented before any compounding personnel begin to prepare CSPs. |
| † Compounding personnel shall complete didactic training, pass written competence assessments, undergo skill assessment using observational audit tools, and media-fill testing. |
| † Media-fill testing of aseptic work skills shall be performed initially before beginning to prepare CSPs and at least annually thereafter for low- and medium-risk level compounding; and semiannually for high-risk level compounding. |
| † Compounding personnel who fail written tests, observational audits, or whose media-fill test vials have one or more units showing visible microbial contamination, shall be reinstructed and re-evaluated by expert compounding personnel to ensure correction of all aseptic work practice deficiencies. |
| † Compounding personnel shall pass all evaluations prior to resuming compounding of sterile preparations. |
| † Compounding personnel must demonstrate proficiency of proper hand hygiene, garbing, and consistent cleaning procedures in addition to didactic evaluation and aseptic media fill. |
| † Cleaning and disinfecting procedures performed by other support personnel shall be thoroughly trained in proper hand hygiene, and garbing, cleaning, and disinfection procedures by a qualified aseptic compounding expert. |
| † Support personnel shall routinely undergo performance evaluation of proper hand hygiene, garbing, and all applicable cleaning and disinfecting procedures conducted by a qualified aseptic compounding expert. |
| Competency Evaluation of Garbing and Aseptic Work Practices |
| † Compounding personnel shall be evaluated initially prior to beginning compounding CSPs and whenever an aseptic media fill is performed using a Sample Form for Assessing Hand Hygiene and Garbing Related Practices of Compounding Personnel and the personnel glove fingertip sampling procedures. |
| Aseptic Work Practice Assessment and Evaluation via Personnel Glove Fingertip Sampling |
| † Monitoring of compounding personnel glove fingertips shall be performed for all CSP risk level compounding. |
| † Glove fingertip sampling shall be used to evaluate the competency of personnel in performing hand hygiene and garbing procedures in addition to educating compounding personnel on proper work practices. |
| † All personnel shall demonstrate competency in proper hand hygiene and garbing procedures in addition to aseptic work practices. |
| † Sterile contact agar plates shall be used to sample the gloved fingertips of compounding personnel after garbing to assess garbing competency and after completing the media-fill preparation. |
| † Gloves shall not be disinfected with sterile 70% IPA immediately prior to sampling. |
| Garbing and Gloving Competency Evaluation |
| † Compounding personnel shall be visually observed during the process of performing hand hygiene and garbing procedures. |
| † The visual observation shall be documented on a Sample Form for Assessing Hand Hygiene and Garbing Related Practices of Compounding Personnel and maintained to provide a permanent record of and long-term assessment of personnel competency. |
| Gloved Fingertip Sampling |
| † Immediately after the compounder completes the hand hygiene and garbing procedure, the evaluator shall collect a gloved fingertip and thumb sample from both hands of the compounder onto appropriate agar plates by lightly pressing each finger tip into the agar. |
| † The plates shall be incubated for the appropriate incubation period and at the appropriate temperature. |
| † All employees shall successfully complete an initial competency evaluation and gloved fingertip/thumb sampling procedure (0 cfu) no less than three times before initially being allowed to compound CSPs for human use. |
| † After completing the initial gowning and gloving competency evaluation, re-evaluation of all compounding personnel shall occur at least annually for low- and medium-risk level CSPs and semiannually for high-risk level CSPs before being allowed to continue compounding CSPs. |
| † Gloves shall not be disinfected with sterile 70% IPA prior to testing. |
| † The sampled gloves shall be immediately discarded and proper hand hygiene performed after sampling. The nutrient agar plates shall be incubated as stated below. |
| † The cfu action level for gloved hands shall be based on the total number of cfu on both gloves and not per hand. |
| ‡ Results should be reported separately as number of cfu per employee per hand (left hand, right hand). |
| Incubation Period |
| † At the end of the designated sampling period, the agar plates are recovered, covers secured, inverted and incubated at a temperature and for a time period conducive to multiplication of microorganisms. Trypticase soy agar (TSA) with lecithin and polysorbate 80 shall be incubated at 35 |
| Aseptic Manipulation Competency Evaluation |
| † All compounding personnel shall have their aseptic technique and related practice competency evaluated initially during the media-fill test procedure and subsequent annual or semiannual media-fill test procedures on the Sample Form for Assessing Aseptic Technique and Related Practices of Compounding Personnel. |
| Media-Fill Test Procedure |
| † The skill of personnel to aseptically prepare CSPs shall be evaluated using sterile fluid bacterial culture media-fill verification. |
| † Media-filled vials shall be incubated within a range of 35 |
| Surface Cleaning and Disinfection Sampling and Assessment |
| † Surface sampling shall be performed in all ISO classified areas on a periodic basis and can be accomplished using contact plates and/or swabs and shall be done at the conclusion of compounding. |
| † Locations to be sampled shall be defined in a sample plan or on a form. |
| Cleaning and Disinfecting Competency Evaluation |
| † Compounding personnel and other personnel responsible for cleaning shall be visually observed during the process of performing cleaning and disinfecting procedures during initial personnel training on cleaning procedures, changes in cleaning staff and at the completion of any Media-Fill Test Procedure. |
| † Visual observation shall be documented on a Sample Form for Assessing Cleaning and Disinfection Procedures and maintained to provide a permanent record of, and long-term assessment of, personnel competency. |
| Surface Collection Methods |
| † Immediately after sampling a surface with the contact plate, the sampled area shall be thoroughly wiped with a non-shedding wipe soaked in sterile 70% IPA. |
| ‡ Results should be reported as cfu per unit of surface area. |
| Action Levels, Documentation, and Data Evaluation |
| † Environmental sampling data shall be collected and reviewed on a routine basis as a means of evaluating the overall control of the compounding environment. |
| † If an activity consistently shows elevated levels of microbial growth, competent microbiology personnel shall be consulted. |
| † An investigation into the source of the contamination shall be conducted. |
| † When gloved fingertip sample results exceeds action levels after proper incubation, a review of hand hygiene and garbing procedures as well as glove and surface disinfection procedures and work practices shall be performed and documented. |
| ‡ Any cfu count that exceeds its respective action level should prompt a re-evaluation of the adequacy of personnel work practices, cleaning procedures, operational procedures, and air filtration efficiency within the aseptic compounding location. |
| SUGGESTED STANDARD OPERATING PROCEDURES |
| † All facilities are required to have these, and they must include at least the items enumerated in this section. |
| FINISHED PREPARATION RELEASE CHECKS AND TESTS |
| Inspection of Solution Dosage Forms and Review of Compounding Procedures |
| † Review procedures and documents to ensure sterility, purity, correct identities and amounts of ingredients, and stability. |
| † Visually inspect for abnormal particulate matter and color, and intact containers and seals. |
| Sterility Testing |
| † High-risk level CSPs prepared in batches of more than 25 identical containers, or exposed longer than 12 hours at 2 |
| Bacterial Endotoxin (Pyrogen) Testing |
| † High-risk level CSPs, excluding those for inhalation and ophthalmic administration, prepared in batches of more than 25 identical containers, or exposed longer than 12 hours at 2 |
| Identity and Strength Verification of Ingredients |
| † Written procedures to verify correct identity, quality, amounts, and purities of ingredients used in CSPs. |
| † Written procedures to ensure labels of CSPs contain correct names and amounts or concentrations of ingredients, total volumes, beyond-use dates, storage conditions, and route(s) of administration. |
| STORAGE AND BEYOND-USE DATING |
| Determining Beyond-Use Dates |
| † Use the general criteria in USP |
| MAINTAINING STERILITY, PURITY, AND STABILITY OF DISPENSED AND DISTRIBUTED CSPs |
| † Written procedures for proper packaging, storage, and transportation conditions to maintain sterility, quality, purity, and strength of CSPs. |
| Redispensed CSPs |
| † When sterility, and acceptable purity, strength, and quality can be ensured. |
| † Assignment of sterility storage times and stability beyond-use dates that occur later than those of originally dispensed CSPs must be based on results of sterility testing and quantitative assay of ingredients. |
| Packaging and Transporting CSPs |
| † Packaging maintains physical integrity, sterility, stability, and purity of CSPs. |
| † Modes of transport that maintain appropriate temperatures and prevent damage to CSPs. |
| PATIENT OR CAREGIVER TRAINING |
| † Multiple component formal training program to ensure patients and caregivers understand the proper storage, handling, use, and disposal of CSPs. |
| PATIENT MONITORING AND ADVERSE EVENTS REPORTING |
| † Written standard procedures describe means for patients to ask questions and report concerns and adverse events with CSPs, and for compounding supervisors to correct and prevent future problems. |
| ‡ Adverse events and defects with CSPs reported to FDA's MedWatch and USP's MEDMARX programs. |
Appendix II. Common Disinfectants Used in Health Care for Inanimate Surfaces and Noncritical Devices, and Their Microbial Activity and Properties1
| Chemical Category of Disinfectant | |||||||
| Isopropyl alcohol | Accelerated Hydrogen peroxide3 | Quaternary Ammonium (e.g., dodecyl dimethyl ammonium chloride) | Phenolics | Chlorine (e.g., sodium hypochlorite) | Iodophors (e.g., povidone-iodine) | ||
| Concentration Used | 60–95% | 0.5% | 0.4–1.6% aq | 0.4–1.6% aq | 100–5000 ppm | 30–50 ppm | |
| Microbial Inactivation | Bacteria | + | + | + | + | + | + |
| Lipophilic viruses | + | + | + | + | + | + | |
| Hydrophilic viruses | ± | + | ± | ± | + | ± | |
| M.tuberculosis | + | + | ± | + | + | ± | |
| Mycotic agents (fungi) | + | + | + | + | + | ± | |
| Bacterial Spores | + | ||||||
| Important Chemical & Physical Properties | Shelf life >1 week | + | + | + | + | + | + |
| Corrosive or deleterious effects | ± | ± | ± | ||||
| Non-evaporable residue | + | + | + | ||||
| Inactivated by organic matter | + | ± | + | ± | + | + | |
| Skin irritant | ± | + | + | + | ± | ||
| Eye irritant | + | + | + | + | + | ||
| Respiratory irritant | + | ||||||
| Systemic toxicity | + | + | + | + | + | ||
| Key to abbreviation and symbols: aq = diluted with water; ppm = parts per million; + = yes; |
|||||||
|
1
Modified from World Health Organization, Laboratory Bio Safety Manual 1983 and Rutala WA, “Antisepsis, disinfection and sterilization in the hospital and related institutions,” Manual of Clinical Microbiology, American Society for Microbiology, Washington, DC, 1995, pages 227-245.
|
|||||||
|
2
Inactivation of the most common microorganisms (i.e., bacteria) occurs with a contact time of
|
|||||||
|
3
Accelerated hydrogen peroxide is a new generation of hydrogen peroxide-based germicides in which the potency and performance of the active ingredient have been enhanced and accelerated through the use of appropriate acids and detergents.
|
|||||||
Appendix III. Sample Form for Assessing Hand Hygiene and Garbing Related Practices of Compounding Personnel
| Printed name and position/title of person assessed: | ||||||
| Name of facility or location: | ||||||
| Hand Hygiene and Garbing Practices: The qualified evaluator will check each space for which the person being assessed has acceptably completed the described activity, prints N/A if the activity is not applicable to the assessment session or N/O if the activity was not observed.* | ||||||
| Presents in a clean appropriate attire and manner. | ||||||
| Wears no cosmetics or jewelry (watches, rings, earrings, etc. piercing jewelry included) upon entry into ante-areas. | ||||||
| Brings no food or drinks into or stored in the ante-areas or buffer areas. | ||||||
| Is aware of the line of demarcation separating clean and dirty sides and observes required activities. | ||||||
| Dons shoe covers or designated clean-area shoes one at a time, placing the covered or designated shoe on clean side of the line of demarcation, as appropriate. | ||||||
| Dons beard cover if necessary. | ||||||
| Dons head cover assuring that all hair is covered. | ||||||
| Dons face mask to cover bridge of nose down to include chin. | ||||||
| Performs hand hygiene procedure by wetting hands and forearms and washing using soap and warm water for at least 30 seconds. | ||||||
| Dries hands and forearms using lint-free towel or hand dryer. | ||||||
| Selects the appropriate sized gown examining for any holes, tears, or other defects. | ||||||
| Dons gown and ensures full closure. | ||||||
| Disinfects hands again using a waterless alcohol-based surgical hand scrub with persistent activity and allows hands to dry thoroughly before donning sterile gloves. | ||||||
| Dons appropriate sized sterile gloves ensuring that there is a tight fit with no excess glove material at the fingertips. | ||||||
| Examines gloves ensuring that there are no defects, holes, or tears. | ||||||
| While engaging in sterile compounding activities, routinely disinfects gloves with sterile 70% IPA prior to work in the direct compounding area (DCA) and after touching items or surfaces that may contaminate gloves. | ||||||
| Removes PPE on the clean side of the ante-area. | ||||||
| Removes gloves and performs hand hygiene. | ||||||
| Removes gown and discards it, or hangs it on hook if it is to be reused within the same work day. | ||||||
| Removes and discards mask, head cover, and beard cover (if used). | ||||||
| Removes shoe covers or shoes one at a time, ensuring that uncovered foot is placed on the dirty side of the line of demarcation and performs hand hygiene again. (Removes and discards shoe covers every time the compounding area is exited). | ||||||
| *The person assessed is immediately informed of all unacceptable activities (i.e., spaces lacking check marks, N/A, or N/O) and shown and informed of specific corrections. | ||||||
| Signature of Person Assessed | Printed Name | Date | ||||
Appendix IV. Sample Form for Assessing Aseptic Technique and Related Practices of Compounding Personnel
| Printed name and position/title of person assessed: | ||||||
| Name of facility or location: | ||||||
| Aseptic Technique, Safety, and Quality Assurance Practices: The qualified evaluator checks each space for which the person being assessed has acceptably completed the described activity, prints N/A if the activity is not applicable to the assessment session or N/O if the activity was not observed.* | ||||||
| Completes the Hand Hygiene and Garbing Competency Assessment Form. | ||||||
| Performs proper hand hygiene, garbing, and gloving procedures according to SOPs. | ||||||
| Disinfects ISO Class 5 device surfaces with an appropriate agent. | ||||||
| Disinfects components/vials with an appropriate agent prior to placing into ISO Class 5 work area. | ||||||
| Introduces only essential materials in a proper arrangement in the ISO Class 5 work area. | ||||||
| Does not interrupt, impede, or divert flow of first-air to critical sites. | ||||||
| Ensures syringes, needles, and tubing remain in their individual packaging and are only opened in ISO Class 5 work area. | ||||||
| Performs manipulations only in the appropriate DCA of the ISO Class 5 device. | ||||||
| Does not expose critical sites to contact contamination or worse than ISO Class 5 air. | ||||||
| Disinfects stoppers, injection ports, and ampul necks by wiping with sterile 70% IPA and allows sufficient time to dry. | ||||||
| Affixes needles to syringes without contact contamination. | ||||||
| Punctures vial stoppers and spikes infusion ports without contact contamination. | ||||||
| Labels preparation(s) correctly. | ||||||
| Disinfects sterile gloves routinely by wiping with sterile 70% IPA during prolonged compounding manipulations. | ||||||
| Cleans, sets up, and calibrates automated compounding device (e.g., “TPN compounder”) according to manufacturer's instructions. | ||||||
| Disposes of sharps and waste according to institutional policy or recognized guidelines. | ||||||
| *The person assessed is immediately informed of all unacceptable activities (i.e., spaces lacking check marks, N/A, or N/O) and shown and informed of specific corrections. | ||||||
| Signature of Person Assessed | Printed Name | Date | ||||
Appendix V. Sample Form for Assessing Cleaning and Disinfection Procedures
| Printed name and position/title of person assessed: | ||||||
| Name of facility or location: | ||||||
| Cleaning and Disinfection Practices: The qualified evaluator will check each space for which the person being assessed has acceptably completed the described activity, prints N/A if the activity is not applicable to the assessment session or N/O if the activity was not observed.* | ||||||
| Daily Tasks: | ||||||
| Prepares correct concentration of disinfectant solution according to manufacturer's instructions. | ||||||
| Uses appropriately labeled container for the type of surface to be cleaned (floor, wall, production bins, etc.). | ||||||
| Documents disinfectant solution preparation. | ||||||
| Follows garbing procedures when performing any cleaning activities. | ||||||
| At the beginning of each shift, cleans all ISO Class 5 devices prior to compounding in the following order: walls, IV bar, automated compounders, and work surface. | ||||||
| Uses a lint free wipe soaked with sterile 70% IPA or other approved disinfectant solution and allows to dry completely. | ||||||
| Removes all compounder components and cleans all ISO Class 5 areas as stated above at the end of each shift. | ||||||
| Cleans all counters and easily cleanable work surfaces. | ||||||
| Mops floors, using the mop labeled “floors”, starting at the wall opposite the room entry door; mops floor surface in even strokes toward the operator. Moves carts as needed to clean entire floor surface. Use of a microfiber cleaning system is an acceptable alternative to mops. | ||||||
| In the ante-area, cleans sink and all contact surfaces; cleans floor with a disinfectant solution or uses microfiber cleaning system. | ||||||
| Monthly Tasks: | ||||||
| Performs monthly cleaning on a designated day. Prepares a disinfectant solution as stated in daily tasks that is appropriate for the surfaces to be cleaned. | ||||||
| Cleans buffer area and ante-area ceiling, walls, and storage shelving with a disinfectant solution and a mop or uses a microfiber cleaning system. | ||||||
| Once ISO Class 5 area is clean, cleans compounding room ceiling, followed by walls and ending with the floor. Uses appropriate labeled mops or microfiber cleaning system. | ||||||
| Cleans all buffer area totes and storage shelves by removing contents and using a germicidal detergent soaked lint free wipe, cleans the inside surfaces of the tote and then the entire exterior surfaces of the tote. Allows totes to dry. Prior to replacing contents into tote, wipes tote with sterile 70% IPA to remove disinfectant residue. Uses new wipe as needed. | ||||||
| Cleans all buffer area carts by removing contents and using germicidal detergent soaked lint free wipe, cleans all carts starting with the top shelf and top of post, working down to wheels. Cleans the under side of shelves in a similar manner. Uses a new wipe for each cart. Allows to dry. Wipes carts with sterile 70% IPA wetted lint-free wipe to remove any disinfectant residue. Uses new wipe as needed. | ||||||
| Cleans buffer area chairs, the interior and exterior of trash bins, and storage bins using disinfectant solution soaked lint free wipe. | ||||||
| Documents all cleaning activities as to who performed such activities with date and time noted. | ||||||
| *The person assessed is immediately informed of all unacceptable activities (i.e., spaces lacking check marks, N/A, or N/O) and shown and informed of specific corrections. | ||||||
| Signature of Person Assessed | Printed Name | Date | ||||
1
See American Society of Heating, Refrigerating and Air-Conditioning Engineers, Inc. (ASHRAE), Laboratory Design Guide.
2
CETA Applications Guide for the Use of Compounding Isolators in Compounding Sterile Preparations in Healthcare Facilities, CAG-001-2005, Controlled Environment Testing Association (CETA), November 8, 2005.
3
U.S. Food and Drug Administration, Guidance for Industry, Sterile Drug Products Produced by Aseptic Processing—Current Good Manufacturing Practice, September 2004.
4
Guidelines for Environmental Infection Control in Health-Care Facilities, Recommendations of CDC and the Healthcare Infection Control Practices Advisory Committee (HICPAC), MMWR, vol. 52, no. RR-10, June 6, 2003, figure 3, pg. 12.
5
NSF/ANSI 49.
6
ISO 14644-4:2001 Cleanrooms and associated controlled environments—Design, construction, and start-up, Case Postale 56, CH-1211 Geneve 20, Switzerland, tel. +41 22 749 01 11.
7
By definition (IEST RP CC 001.4), HEPA filters are a minimum of 99.97% efficient when tested using 0.3-µm thermally generated particles and a photometer or rated at their most penetrating particle size using a particle counter.
8
Sample procedures are detailed in CETA Applications Guide CAG-002-2006–section 2.09.
9
Controlled Environment Testing Association, 1500 Sunday Drive, Ste. 102, Raleigh, NC 27607; www.CETAinternational.org.
10
Agalloco J, Akers JE. Aseptic Processing: A Vision of the Future. Pharmaceutical Technology, 2005. Aseptic Processing supplement, s16.
11
Eaton T. Microbial Risk Assessment for Aseptically Prepared Products. Am Pharm Rev. 2005; 8 (5, Sep/Oct): 46–51.
12
Guideline for Hand Hygiene in Health care Settings, MMWR, October 25, 2002, vol. 51, No. RR-16 available on the Internet at http://www.cdc.gov/handhygiene/.
13
The use of additional resources, such as the Accreditation Manual for Home Care from the Joint Commission on Accreditation of Healthcare Organizations, may prove helpful in the development of a QA plan.
Auxiliary Information— Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
| General Chapter | Claudia C. Okeke, Ph.D.
Scientific Fellow 1-301-816-8345 |
(SCC05) Sterile Compounding 05 |
USP32–NF27 Page 318
Pharmacopeial Forum: Volume No. 32(3) Page 852