MANUFACTURING OF CELL THERAPY PRODUCTS
Introduction
Cell processing for cell therapy applications is a unique form of biologics manufacturing that relies on maintenance of stringent work practices designed to ensure product consistency and prevent contamination by microorganisms or by another patient's cells. Hallmarks of this unique form of manufacturing can include products with limited shelf lives, the need for rigorous control during manual processing steps, a manufacturing environment in which many product lots are simultaneously processed and assembled, raw materials that may or may not be part of the final product, and numerous pieces of processing equipment. By its very nature, cell processing requires a number of operations and manipulations by individuals well trained in aseptic processing techniques. The technical competence of the personnel is particularly crucial to product safety and efficacy with this form of manufacturing. Procedures involving lot segregation, line clearance, and operational discipline must be developed to decrease the chance of mix-up of patient-specific lots.
The degree of control required for cell processing operations is highly dependent upon a number of factors, including the complexity of an aseptic manufacturing process, the primary site of manufacturing, and the mode of administration of the cell product to the patient. Manufacturing processes that involve open manipulation of the cells even in a biological safety cabinet are at greater risk of contamination than the processes done in closed bioreactors or intravenous transfer bag systems that use sterile connection devices and tube-sealing devices. Clean rooms and biological safety cabinets are essential components for processes that involve open manipulations or for patient-specific products. The controlled environment of a carefully designed, constructed, validated, and maintained clean room will minimize the risks of environmental contamination during aseptic processing and decrease the possibility of cross-contamination of patient-specific products. Processes that utilize closed systems do not require clean room environments.
Procurement of Source Material
A variety of human- and animal-derived tissues, which can also include whole organs, serve as sources of cells for cell therapy products. Examples include skin, muscle, cartilage, bone, neural tissue, bone marrow, blood vessels, parenchymal cells from organs such as the liver, pancreas, and adrenal glands, and stem cells from adult and fetal tissues. A few general principles in the sourcing of these tissues are as follows: (1) systems must be developed so as to allow the material to be traced back to the donor; (2) steps must be taken to prevent the transmission of an infectious disease from the donor to the recipient; and (3) adherence to aseptic procedures during procurement and initial processing are necessary to ensure the safety of the final product because terminal sterilization of cells is not possible.
human tissue
Human-derived tissues may be sourced from normal healthy donors, cadaveric donors, or diseased patients, such as those with cancer. Applicable guidelines and standards for the procurement of human tissue are available from the American Association of Tissue Banks (AATB) and the FDA. Additionally, the federal policy in 45 CFR Part 46 is applicable to all federal or federally supported research. This policy requires that a certified institutional review board review and approve use of any tissue taken from a live human donor. The policy also includes special considerations for research on prisoners, children, and pregnant women or research in other areas involving gestational tissue. In all cases, appropriate written consent must be obtained from the donor or the donor's next of kin, describing which tissue is being procured and for what use it is intended. The donor must meet established guidelines for donor suitability and be tested for the infectious diseases listed in Table 4. The medical history of the donor must be reviewed to ensure the absence of signs and symptoms of these diseases and to rule out issues and behaviors that increase the risk of exposure to such diseases.
Human tissue should be obtained under environmental conditions and controls that provide a high degree of assurance for aseptic recovery. Standard hospital operating room practices are applicable for tissues requiring dissection and surgical procurement. The air quality provided in a typical limited-access operating room is adequate for such procedures. Procurement personnel must be appropriately trained in all aspects of tissue recovery, such as surgical scrubbing, gowning, operating room behavior, anatomy, surgical site preparation, and antisepsis. Special care is required when tissue or organ procurement requires extensive manipulation of the bowel and when sharp dissection may result in the inadvertent puncture of the bowel. Tissue that contains microbial flora (for instance, skin) at the time of procurement can be adequately disinfected by using antimicrobial or bactericidal agents and extensive scrubbing.
Table 4. Infectious Disease Testing for Human Cells and Tissues Used in Cell Therapy Products
Infectious Disease Testing for Human Cells and Tissues Used in Cell Therapy Products
| Testing* | ||||||
| Cell Type | HIV 1, 2 | Hepatitis C | Hepatitis B | HTLV | Cytomegalovirus | Treponema pallidum |
| Autologous stem cells | R | R | R | R | ||
| Other autologous tissue | R | R | R | |||
| Allogeneic stem cells from family-related donors | X | X | X | X | X | X |
| Other allogeneic tissue | X | X | X | X | X | X |
| X—required | ||||||
| R—recommended; the labeling stating “tested negative” or “not tested for biohazards” may be required | ||||||
|
*
For autologous or allogeneic cord blood donors or fetal tissue, a mother's sample may be used for testing.
|
||||||
human blood and bone marrow
Hematopoietic progenitor cells represent one of the most extensively used cell sources in the field of human transplantation. These cells can be collected from the bone marrow, peripheral blood, placental umbilical cord blood, or fetal liver. The source of cells is somewhat dependent upon the patient, the disease, and the clinical protocol. Regardless of the cell source, methods for processing the cells are similar.
Human-derived blood cells and bone marrow cells may be sourced from normal, healthy donors or patients with hematological disorders. Applicable guidelines and standards for the collection and processing of these materials have been published by the American Association of Blood Banks (AABB), the Foundation for the Accreditation of Hematopoietic Cell Therapy, the National Marrow Donor Registry (NMDR), and the FDA. Similar issues regarding consent, infectious disease testing, and donor medical history apply in the sourcing of blood- or bone marrow-derived cells for allogeneic transplants. In cases where these cells will be subjected to selection, expansion, genetic manipulation, or other complex processing procedures, the testing outlined in Table 4 should be followed.
Bone marrow for clinical use is harvested predominantly by percutaneous needle aspiration of the anterior or posterior iliac crests or the sternum. Standard hospital operating room practices are employed by specially trained personnel. Plastic syringes and commercially available aspiration needles are used to draw 3- to 5-mL volumes of marrow from each site of penetration. The material is transferred to a sterile, balanced salt solution or tissue culture medium containing sufficient anticoagulant, such as heparin, to prevent clotting. Removal of bone spicules may be accomplished by passing the material through stainless steel mesh screens or collection kits consisting of sterile, plastic collection bags with in-line filters having about a 200-µm porosity. The volume of marrow collected is dependent upon the body weights and other characteristics of both the donor and the recipient. The maximum volume to be harvested from a donor is about 10 to 15 mL per kg of body weight.
Circulating hematopoietic, peripheral blood progenitor cells (PBPCs) comprise a small population of peripheral blood mononuclear cells that can be utilized in place of or in addition to bone marrow. PBPCs are collected by apheresis, a procedure by which donor blood is withdrawn from a vein and separated ex vivo into some or all of its component parts. One or more of the components are retained as the harvest and the remaining parts are returned to the donor. Conditioning of the donor may enrich the number of circulating PBPCs in the harvest. Examples of such conditioning include collection during recovery from myelosuppressive chemotherapy and administration of hematopoietic growth factors, such as granulocyte colony-stimulating factor (G-CSF) and granulocyte–macrophage colony-stimulating factor (GM-CSF), or steroids. Collections are also improved by increasing the frequency or volume of apheresis. Apheresis requires one or two large-bore peripheral venous catheters in the upper extremities or a single large-bore, thick-walled, central venous double or triple lumen catheter (Mahurkur type). Two types of apheresis technology are available: the discontinuous-flow cell separators (Haemonetics) and the continuous-flow systems (COBE or Fenwall). Anticoagulation for normal to high flow rates is with a citrate-based material. In a closed system, the risk of contamination is low. The procedure is generally performed by trained, dedicated staff in a blood bank or in a donor center associated with a blood bank.
Placental and umbilical cord blood provides a third source of hematopoietic progenitor cells. Compared to bone marrow and PBPCs, the stem cells of placental and umbilical cord blood have a higher proliferative and self-renewal capacity. Volume of collection and thus cell number are limited and depend upon timing and the presence of a dedicated team of personnel. Collections are made during the third stage of labor. Typically, a closed method of collection is employed and involves cannulation or puncture of the umbilical vein with subsequent collection into plastic syringes or blood collection bags containing citrate-based anticoagulant. The procedure is performed in a controlled-access room away from the site of birth. Cellular content of the collection includes large numbers of erythrocytes, leukocytes, platelets, and target mononuclear cells. An open collection technique, which involves drainage of the blood by gravity from the cut end of the cord into sterile tubes containing anticoagulant, does not afford the same aseptic assurance level as the above-mentioned technique.
A major area of concern with the use of placental and umbilical cord blood relates to potential risks of unknown genetic disorders that may be transmitted to the recipient. Donor suitability is established by the usual infectious disease screening of the mother and the completion of a medical questionnaire. The donation remains anonymous and without any long-term follow-up of the child.
animal tissue
The major area of concern with the use of animal tissue relates to the known and unknown risks of potential infectious disease transmission to humans, and as such, the transplantation of animal cells raises unique public health concerns. Introduction of xenogeneic infectious agents into and propagation through the general human population is a risk that must be addressed. Draft Public Health Service (PHS) Guideline on Infectious Disease Issues in Xenotransplantation (August 1996), and any other related regulatory documents that are generated as this field advances, must be consulted when developing xenotransplant cell therapy products. Developers of such products should understand that the product recipients will be subjected to a high level of scrutiny (for instance, clinical and laboratory surveillance or registry in xenotransplantation databases) because of the above-mentioned public health concerns.
The use of animal tissue in the manufacture of cell therapy products requires that the tissue be sourced in a controlled and documented manner and from animals bred and raised in captivity in countries or geographic regions that have appropriate national health status, disease prevention, and control systems. In addition, the care and use of animals should be approved by a certified institutional animal care and use committee. Donor animals must have documented lineage, be obtained from closed herds or colonies, and be under health maintenance and monitoring programs. The facility for housing these animals should be USDA certified (large vertebrate animals) or Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) certified (small vertebrate animals) and should meet the recommendations stated in the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996), which can be obtained from the AAALAC. Such facility should be staffed with veterinarians and other trained personnel who will ensure animal health and disease prevention. The procedures employed in the facility should be documented and records should be kept. Health maintenance and monitoring programs are based on standard veterinary care for the species and include physical examinations, monitoring, laboratory diagnostic tests, and vaccinations. Use of a stepwise batch or all-in–all-out method of movement of source animal through the facility, rather than the continuous replacement movement, is recommended. It allows the decontamination of the facility prior to the introduction of the new set of animals, thereby reducing the chance of disease transmission. Feed components should be documented and should exclude, whenever possible, recycled or rendered materials that may have been associated with the transmission of prior-associated diseases.
To provide a high degree of assurance of product safety, screening of donors and of tissues derived from these donors should be performed at several stages throughout the process to rule out the presence of microbial agents. These control tests should utilize assays that are sufficiently sensitive and specific to detect bacteria, mycoplasma, fungi, or viruses of interest. Donor animals can be screened for certain diseases prior to donation of tissue by applying a variety of serological monitoring tests. Tissues can be subjected to a panel of tests including, but not limited to, the following:
- test for sterility;
- test for mycoplasma;
- test for cultivable viruses in vitro;
- test for unknown viruses by inoculation of various laboratory animals;
- tests for xenotropic endogenous retroviruses and other animal retroviruses by in vitro cocultivation techniques, biochemical methods (for instance, to detect viral reverse transcriptase), and molecular biology assays (such as PCR assay for viral genomic sequence detection); and
- direct detection or observation methods such as electron microscopy, detection of specific viral antigens by fluorescent antibody microscopy, or enzyme immunoassay methods.
Most of these tests are addressed under Analytical Methodologies or under Biotechnology-Derived Articles  1045
1045 and Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin 1050. Post-tissue retrieval necropsies, sentinel animal programs, and archival storage of donor organs, tissues, blood, and other specimens are additional components of the overall program to ensure the safety of animal tissue for use in cellular therapeutic applications.
and Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin 1050. Post-tissue retrieval necropsies, sentinel animal programs, and archival storage of donor organs, tissues, blood, and other specimens are additional components of the overall program to ensure the safety of animal tissue for use in cellular therapeutic applications.
Most of the same aseptic procurement issues apply to animal tissue and to human tissue. Again, the tissue should be obtained under environmental conditions and controls that provide a high degree of assurance of aseptic recovery. Specifically designed procurement facilities, usually closely associated with the animal holding facility, are typically employed. These facilities have specific attributes and design features that may not be available or applicable in the hospital operating-room setting. Such features include the following: (1) staging of various events, such as shaving, sedation, and operating-room preparation, in different rooms that are often separated with air locks for environmental control; (2) high-efficiency particulate air (HEPA) filtration; (3) adjacent but separate facilities for further tissue processing; and (4) dedicated areas for carcass removal. The issues regarding the training of personnel, bowel manipulation and puncture, and disinfection that are applicable to human tissues apply to the surgical procurement of tissue from animals as well (see Human Tissue).
Cell Isolation and Selection
general considerations
The general principles for processing human and animal tissues following aseptic procurement are independent of the tissue source. The manufacture of cell products may occur at a clinical site or at a central cell-processing facility. Sites involved in cell processing should ensure reproducibility and safety of the manufactured products through appropriate QC and QA programs.
Regardless of the location, processing should occur in a dedicated area physically separated from the site of procurement. To the greatest extent possible, the facility design and processing procedures should be consistent with those provided by the FDA's Guidelines on Sterile Drug Products Produced by Aseptic Processing (June 1987), or provided under Pharmaceutical Compounding—Sterile Preparations 797 for processes involving open manipulation. Generally, this requires that properly trained and outfitted processing staff handle blood or tissue samples in a critical zone supplied with class 100 HEPA-filtered air, which is provided by a biological safety cabinet located in a controlled clean room supplied with class 10,000 HEPA-filtered air. The facility and processing areas should be monitored for air quality in a manner that provides a high level of process asepsis. For guidance in this area, see Microbiological Evaluation of Clean Rooms and Other Controlled Environments 1116. The material should be packaged in sterile, leak-proof containers and transported from the procurement area to the processing area under controlled conditions that maintain cell viability. The fluid medium in which the specimens are bathed during transportation should be optimized to maintain cell and tissue viability. This transport medium can be supplemented with antibiotics. If so, the antibiotic levels in process buffers are decreased and eventually eliminated during subsequent processing steps, so that antibiotics are not present in the final cellular product. In the case of blood products or tissues containing substantial amounts of blood, the transport media or buffered electrolyte solution should contain an anticoagulant such as heparin or a citrate-based material.
isolation
Solid organs or tissues are usually dissected to expose a desired region. This material may be used as is for transplantation or it may be processed further. If multicellular organoids (for instance, islets of Langerhans) or single-cell suspensions are desired, the tissue may be subjected to mechanical or enzymatic disaggregation. Physical disaggregation may be accomplished through the use of instruments that impart high shear forces on the material (namely, to homogenize) or break the tissue into smaller pieces. Alternatively, the material can be pressed or passed through screens of defined mesh sizes.
Enzymatic digestion of the extracellular connective tissue, which holds cells together within the tissue, is another common method for dissociating solid tissue. Typically, the tissue is minced into small cubes, usually larger than 1 mm3, and incubated in a buffered solution containing a digestive enzyme. Alternatively, the intact organ is infused with a solution to rinse the blood from the tissue followed by the enzymatic solution that aids the digestion. Various enzymes are used to accomplish this. Examples include collagenase, trypsin, elastase, hyaluronidase, papain, and chymotrypsin. Enzymes with nuclease activity, such as deoxyribonuclease, may be added to digest nucleic acids released from damaged cells, preventing excessive cell clumping. At the end of the incubation process, the cell suspension may be subjected to a mild pumping action to further break up multicellular clusters into those of desired size or composition. Enzymatic and physical disaggregation methods are often combined to achieve the desired result.
Because cells isolated from blood and bone marrow products are inherently cell suspensions, mechanical manipulation is limited to plasma removal, which is accomplished by centrifugation and physical removal of clots that occurred during transport via 200-µm filtration.
selection
Cell suspensions at this stage may be transferred directly to culture vessels as described for Propagation under Cell Propagation and Differentiation, genetically manipulated as described under Introduction of Genetic Material into Cells, or formulated by various techniques as described under Formulation of Cell Therapy Products. Cell suspensions often consist of a mixture of cell types that may require further processing to isolate a cell population of interest or to decrease the level of an undesirable cell type such as potentially contaminating tumor cells. Various cell isolation and separation techniques exist that provide high yields of pure cell populations.
Each cell type typically possesses specific size and density; therefore, different cell types will sediment at different rates in a centrifugal field or at unit gravity. Cell populations can be selectively sedimented to yield pure fractions by varying the centrifugation forces and the duration of centrifugation. Separation can also be achieved by isopycnic centrifugation, where the cell suspension is centrifuged in a gradient medium that encompasses all of the densities of cells in the sample. In this procedure, the various cell populations sediment to an equilibrium position at the gradient density equal to the density of the cell population. Specifically designed continuous-flow elutriation centrifuges separate cell populations by subjecting a cell suspension to opposite centrifugal and fluid stream forces in a special chamber within the centrifuge rotor mechanism. Cell populations separate within the rotor on the basis of their various sizes and densities, and they are selectively eluted out of the rotor chamber by increasing the fluid stream force. Finally, methods that do not require centrifugation but instead involve the addition of high-density agents, such as hydroxyethyl starch, to the cell suspension will result in cell separation. The mixture is allowed to settle in a tube at unit gravity, resulting in the separation of different cell types based on buoyant density. Concentration and separation procedures such as these frequently result in cell loss due to clumping and aggregation.
Cell separation can also be achieved by applying techniques that take advantage of unique cytological or biochemical characteristics of different cell populations. Soybean agglutinin binds to and agglutinates cells that bear a particular carbohydrate moiety expressed on mature blood cells, but not stem cells, allowing for purification of the stem cells. Lymphocytes possess the CD2 antigen that acts as a receptor for sheep red blood cells. The lymphocytes form rosettes, which then can be separated via differential centrifugation.
Some applications take advantage of the ability of certain cell populations to adhere to the surface of specific solid substrates such as tissue culture plastic, collagen-coated materials, and natural and synthetic polymeric scaffolds. The specifically bound cell type is selectively recovered onto the surface and removed from the initial cell suspension. When placed under the appropriate culture conditions, these cells will multiply and eventually occupy the available surface or void volume of the substrate.
Monoclonal antibodies directed against specific cell surface antigens or receptors can be used for both positive and negative cell selection. For example, a monoclonal antibody–labeled cell population can be removed from the cell suspension immunomagnetically, after exposure to magnetic particles coated with antimonoclonal antibody. The magnetic particles and their bound cells are removed from the cell suspension magnetically. Cells are released from the complex following incubation with reagents, such as specific peptides, that dissociate the monoclonal antibody from the cell. Unlabeled cell suspensions can be poured over or incubated on surfaces such as plastic flasks or microspheres coated with monoclonal antibodies as a means of isolating particular cell populations. In addition, a fluorescence-activated cell sorter (FACS) can be used to separate different cell types by binding antibodies tagged with fluorescent markers to a particular cell type.
Various other techniques purify particular cell populations by destroying unwanted cells present in the mixture. For example, certain cell-bound monoclonal antibodies are able to fix and activate complement, which is added to the cell suspension, resulting in lysis of the cell. Some procedures use cytotoxic agents or mitotic inhibitors to selectively impede or kill unwanted cells in a cell product. These methods typically target an unwanted cell subpopulation with a high growth rate, such as tumor cells. Finally, an antibody can be conjugated to a toxic moiety, such as ricin, allowing delivery of the cytotoxic agent to the targeted cell population. Most of these procedures require several washing steps after the exposure of the cells to the cytotoxic agents to ensure the removal of the dead cells, cell fragments, and cytotoxic agents from the final cell product.
Cell Propagation and Differentiation
propagation
A key issue for cell therapy products is the ability to manufacture and deliver a therapeutically relevant dose of the required cell population to the patient. Depending on the application, the product may be a pure, homogeneous cell type or it may be a mixture of different functional cell types. Many target cell populations are present at low level or low purity in complex primary source tissues. In such cases, production of a therapeutic dose may be achieved only by specific enrichment and propagation of the required cells.
Propagation of cells may occur in suspension culture (for example, T cells or hematopoietic stem and progenitor cells), adherent culture (for example, mesenchymal stem cells, embryonic stem cells, neuronal stem cells, or dermal fibroblasts), or a mixture of both (for example, bone marrow stroma expansion). Numerous devices of varying degrees of sophistication and automation exist for cell culture.
In the simplest iteration, cells can be propagated in tissue culture flasks (T flasks), roller bottles, on polymeric scaffolds, or nonrigid, gas-permeable bags inside regular incubator units that are controlled for temperature, humidity, and gas composition. Multilayered plastic cell factories, cell cubes, and multi-bag systems have been developed that enable expansion, harvesting, and formulation to be carried out in a closed system.
Traditional small-scale fermenter units can be used for expansion of cells in suspension culture. It is also possible to expand adherent cells in such units either by providing a surface for attachment (coated beads or disks) or by adapting the cells to propagate in suspension culture. Some culture systems are specifically designed for the propagation of cells for therapeutic applications. These systems tend to be closed systems that use disposable bioreactor cartridges, such as those made of hollow fiber or molded plastic, in automated processing units with direct control of parameters such as temperature, gas composition, and media perfusion rate. These units can provide a completely automated, closed system for expansion and harvesting. In some cases the automated software is set up for patient–donor tracking and will document culture conditions and manipulations for the entire processing run. These features are useful in the design and implementation of QC product-release testing programs and for the QA documentation of processing runs.
In the case of adherent culture, the cells are usually released from the surface upon which they have expanded. Methods of release include physical agitation, enzymatic cleavage with enzymes such as porcine or bovine trypsin, collagenase, or dextranase, chelation of metal ions (for example, with edetate disodium), and competitive inhibition of adhesion or matrix molecules. As described above, consideration must be given to the source, safety, toxicology, and residual testing for any reagent used to release adherent cells during manufacturing.
Some product-specific systems that do not require the release of adherent cells have been developed. In these systems, the cells are expanded upon a synthetic or natural matrix that is then applied topically (for example, in dermal repair products) or the cells are grown inside or outside of fibers for ex vivo perfusion (for example, hepatocytes in hollow-fiber devices to treat liver disease). In these applications, the matrix and device composition must be biocompatible and, in some cases, biodegradable.
In all of the above systems, standard cell culture parameters must be optimized for maximum process efficiency. Such parameters include composition of cellular source material, initial seeding density, media composition, rate of media exchange, temperature, gas composition, and rate of delivery. Depending on the nature of the product, the potential effect of process parameters on the potency and function of the target cells should be defined.
In closed bioreactor systems, it can be difficult to observe or sample cells so as to determine and control the rate of proliferation and thereby the point of harvest. Measurement of traditional fermentation parameters, such as rate of nutrient usage or production of metabolic products, can provide a surrogate method, amenable to validation, with which to evaluate the rate of proliferation and predict when to harvest the cell product. The relationship of such parameters to the viability, potency, and function of the cell product should be well defined. Postexpansion purification and enrichment of target cells by using methods such as those described above may be required.
differentiation
Some cell therapies require lineage or functional differentiation of the source cells. For example, hematopoietic stem cell expansion processes normally result in products containing a mixture of multipotent stem cells, lineage-committed progenitor cells, and lineage-differentiated cells. The composition of these products can be manipulated by using different combinations of growth factors and cytokines during the expansion process. The inverse is true for processes in which mature cells are de-differentiated to enable them to then be recommitted to a lineage pathway (for example, chondrocytes in cartilage repair).
Specific examples of ex vivo manipulation are the programming of professional antigen-presenting cells, such as dendritic cells and monocytes or macrophages, and the production of antigen-specific T cells to target various specific disease indications. In these applications, the manipulated cells may be engineered to target and attack a specific tumor or tumor cell type, to induce a specific antibody or other cellular response, or to potentially vaccinate a patient. The processes for production of such products can involve one or more exposures of the relevant cells to disease-specific synthetic immunogens (for example, peptides) or natural immunogens (for example, dead tumor cells, viruses, cell membrane fractions, or purified natural molecules) before, after, or during culture expansion. Alternatively, the target cells may be genetically engineered with a specific gene product, such as an HIV-specific receptor. In some applications, relevant cells are cocultured with tumor cells, other diseased cells, or cells producing a transduceable or transfectable gene construct to generate a specifically targeted product.
Again, prior to delivery, the manipulated target cells may require further purification and enrichment by applying the methods described throughout this section. In the case of certain T-cell products, the desired antigen-specific cells can be cloned and then further expanded to provide the therapeutic dose.
Introduction of Genetic Material into Cells
A common extension of cell therapy involves the introduction of genetic material, usually DNA, into cells to alter their pattern of gene expression. For the purpose of this section, it is assumed that the nucleic acid is DNA. Similar scenarios can be applied to RNA or a derivative of DNA, except that the stability and solubility of the particular nucleic acid may dictate modifications of certain steps. This process is often referred to as ex vivo gene therapy, because the cells are removed from the patient or donor and the genetic material is introduced while the cells are outside of the body. Genetically modified cells are then administered to the patient. The genetic material introduced can either cause the expression of new genes and products or cause the inhibition of the expression of already expressed genes and products. The latter represents a type of antisense therapy. The genetic material can be introduced by the same range of reagents that are involved with gene therapy: viral vectors, nucleic acids in a simple formulation (naked DNA), or nucleic acids formulated with agents, such as liposomes, that enhance their ability to penetrate the cell. Most of the steps and considerations discussed above also apply to the ex vivo introduction of genetic material into cells. However, the main goal of ex vivo therapy is to develop robust processes that will work with the majority of patient's or donor's cells. This takes considerably more effort than processes for cell lines.
The method of introduction of new genetic material into cells depends on the biology of the system and the desired stability of gene expression. If a simple retroviral vector such as Molony murine leukemia virus is used for transduction, the cells must be actively dividing because vector DNA is only integrated into the cellular DNA during replication. This usually leads to long-lasting expression of the desired gene product. Adenoviral vectors, naked DNA, or formulated DNA can be introduced into nondividing cells. However, gene expression will be transient, because the introduced DNA will usually be extrachromosomal.
The main challenge is to achieve efficient transduction or transfection, introducing sufficient DNA into the cell before the DNA degrades. In the case of transduction by retroviral vectors, vectors derived from simple retroviruses, cells are stimulated with reagents that cycle them into the S phase (replication) at the time the vector is applied. Most retroviral vectors are stable in cell culture for a period up to a few hours. Because diffusion is minimal, only a small fraction of viral particles will come into contact with cells over this period. The following techniques can be used to increase the number of viral particles that contact the cell in a given time period:
- maximization of viral particle concentration and minimization of the media volume during the transduction step
- multiple applications of the virus
- centrifugation of virus particles onto the cells
- placing of cells on a filter and slow pulling of viral media through the filter
- addition of binding-enhancing polymers to the media.
note—Coculturing of the target cells with the viral producer cells is not recommended. This technique increases the chance of a recombinant event occurring and of the production of RCV. Furthermore, any product for which coculturing is used to transduce the human cells would be considered a xenotransplant if the producer cells were not human. The second cell type, whether human or not, may cause inflammation.
Each of the above techniques has its own set of issues that must be addressed in order to develop a robust process. In technique 1, reduction of the volume during transduction results in rapid exhaustion of the medium; therefore, supplemental medium should be added within a few hours. In technique 2, the cells may no longer be in the correct cell cycle phase during later applications or cells may have become refractory because of unproductive transformation during the prior application. Techniques 3 and 4 can work well on a very small scale, but the number of cells that can be transduced may be insufficient to obtain an efficacious dose. In technique 5, polymers may fail to provide a benefit because virus-binding may involve specific receptors whose surface density may prove to be the limiting factor.
Similar issues and techniques can apply with other viruses or DNA preparations. The issue of slow diffusion is even more marked for the use of DNA preparations. Factors such as the cell type in which the viral vector was produced, the media used for vector production, and the purity of the vector can have a dramatic effect on the efficiency of transduction.
While certain methods may not require cells to be actively cycling, in practice, most processes will require that cells be capable of replication because of the following considerations:
- Safety considerations may dictate that only cells that express the new DNA are returned to the patient, which requires that these cells be selected. As described below, the most common selection method utilizes an antibiotic-resistant gene that is co-introduced with the new genetic material.
- Further propagation may be required to achieve the therapeutic dose of cells.
- Economic, biological, or technical reasons may dictate that the DNA introduction step be carried out at a low cell number and that the desired cell population then be expanded to the required dose.
Therefore, conditions that enable the cell or maintain the cell's ability to proliferate must be developed in almost all cases. The biology of the cells, the available technology, and the process economics will determine whether cells are propagated before, after, or during the introduction of new genetic material. Most processes do in fact expand the population after the introduction of the new gene.
Whether cells that do not productively express the gene can be administered to patients depends on the biology of the application, the dose required versus the handling capability of the manufacturing system, and most importantly, the toxicity of the nonproductive cell population. Selection of the genetically modified cell population is commonly carried out using an antibiotic-resistance marker gene, such as neomycin, which is co-introduced into the cell with the new genetic material. For neomycin selection, cells in culture are treated with the antibiotic G418 at a concentration and for a period that have been shown to kill cells with nonproductive expression, while allowing the productively expressing cells to proliferate. In this manner it is presumed that cells that are resistant to the antibiotic will also express the DNA of interest. The expression should be tested as a lot-release requirement or verified in a series of mock runs. Because most antibiotics decrease cellular proliferation, optimization of the culture media composition may be necessary for efficient selection and propagation of the gene-modified cells.
Following the antibiotic selection step, a second phase of antibiotic-free cell propagation may be required in order to achieve the desired dose and to rinse residual G418 out of the system. The selected medium and the total time that the cells are in culture can be critical to maintaining the desired expression of the original differentiated functions. An additional issue associated with the use of selection markers is that they generally are nonhuman genes. The expression of these genes usually elicits an immune response.
Process development is often carried out with cells from healthy donors. Consideration should be given to the fact that for very sick patients, it can be difficult to obtain healthy cells that can be stimulated to undergo efficient, sustained replication.
Formulation of Cell Therapy Products
suspensions
Formulations for cell therapy products depend upon the desired length of storage and whether the cells are administered as a suspension or in combination with a matrix. Regardless of the route of administration, cells that will be administered as a suspension can be frozen or not frozen. The most common formulation for cells that are cryopreserved is a 5% to 10% solution of dimethyl sulfoxide (DMSO), with or without hydroxyethyl starch (generally 6%), and a plasma protein, such as 4% to 10% human serum albumin, in a balanced salt solution. DMSO prevents dehydration by altering the increased concentration of nonpenetrating extracellular solutions during ice formation at the time of freezing. The high molecular weight polymeric hydroxyethyl solution protects the cells from dehydration as water is incorporated into the extracellular ice crystals. The use of protein often results in maximum recovery and viability of cells after thawing. Serum (5% to 90%) has been used in place of specific proteins. Some cryopreservation formulations are completely free of protein. If the solution contains a buffer, the pH of the buffer should not be affected by changes in temperature. The optimal concentration of cells for cryopreservation depends on the cell type, but it generally ranges from 106 to 107 cells per mL. The purity of the cell population can also affect recovery. For instance, granulocytes can be damaged by the cryopreservative and the cell viability can decrease. These effects are dependent upon the concentration of cryopreservative. Both effects subject the patient to an increased level of infusion-related toxicity, although this is related to the volume administered and the final concentration of the cryopreservative.
Formulations for cell suspensions stored without freezing generally contain cell culture media, often without any protein. Because cells continue to metabolize their media even at the reduced temperatures used for storage, the medium supplies the amino acids and other nutrients that help in maintaining cell viability.
products combined with biocompatible matrices
Many cell therapy products are administered in combination with a biocompatible matrix. For instance, wound healing or skin substitute products contain cells seeded on a matrix. The biochemical and physical structure of the matrix and the method for combining cells with the matrix are specific to the application. Some common examples include the following:
- Cells loaded into a semipermeable membrane device—Usually the pore size of the membrane is large enough to allow the cell-secreted therapeutic factors to pass, but it is small enough to stop immunoglobulins and host cells from making contact with, destroying, or having an immune response to the therapeutic cells. The device can be a single hollow fiber or a semipermeable capsule with cells inside that secrete therapeutic compounds, or it can be part of a larger system of pumps and filters, such as hollow-fiber modules with hepatocytes for the treatment of liver disease.
- Cells seeded onto a three-dimensional matrix and allowed to propagate and form a tissue-like structure—In the resulting product, the cells are oriented in a unique manner that is important for the intended use of the product (for example, skin substitutes). In some cases, mechanical force has been used for proper cell orientation.
-
Cells encapsulated in a gel or cross-linkable polymer solution—The resulting implantable structure can serve as a culture vessel, as a means to shield the cells from the host's immune system, or as a way to mold cells into a defined shape. Some of the polymers used include alginate, hyaluronic acid, collagen, chitin, or synthetic polymers. Encapsulated pancreatic
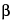 -islet cells have been implanted in patients to treat diabetes. To treat urinary incontinence, chondrocytes have been mixed with alginate to form a structure upon injection.
-islet cells have been implanted in patients to treat diabetes. To treat urinary incontinence, chondrocytes have been mixed with alginate to form a structure upon injection.
- Cells adhered to matrices of defined shape that are then implanted—Some examples include osteogenic precursor cells on matrices of demineralized cadaveric human bone, ceramic hydroxyapatite, ceramic hydroxyapatite–tricalcium phosphate, or biodegradable glass, which can be used in the repair of bone defects.
When manufacturing such products, the primary consideration is the sourcing of a quality matrix material. The matrix material should be biocompatible, should not interfere with cell function, and should not trigger an immune response in the patient. If it is intended that the cells proliferate after loading onto or into the matrix, the matrix and the supporting culture system must allow exchange of nutrients and waste products. Cells may form tissue-like structures under favorable conditions and for those applications where this is required. A thick, impermeable matrix will lead to forming regions of necrotic tissue. Many of these devices are designed so that they can be removed from the patient after a certain period of time.
In all cases where cells are combined with biocompatible matrices, the use of closed systems for the manufacture and the delivery of product is preferable. As cell therapy products of this type can be quite intricate, the manufacturing details for such products are outside the scope of this chapter.