



Add the following:
THEORY
Fluorescence is a two-step process that requires an initial absorption of light followed by emission. [Note—Many terms and variables used in this general chapter are explained in the Appendix: Definitions.
+'&img=../../images/v38332/m3207-f1-pf345.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 1. Fluorescence excitation and emission spectra for fluorescein in borate buffer. The wavelength axis shows excitation and emission wavelengths.
The most common type of fluorescent sample is a dilute, transparent solution that absorbs light following the Beer–Lambert Law and that emits a corresponding fluorescence intensity that is directly proportional to the concentration, the absorptivity, and the fluorescence quantum yield of the fluorescent species or fluorophore. A conventional fluorescence spectrometer has both excitation and emission wavelength selectors. It collects a spectrum by fixing the wavelength of one of the selectors and scanning the other wavelength selector over a range. When the excitation wavelength is fixed and the emission wavelength is scanned, the resulting spectrum is termed an emission spectrum. When the emission wavelength is fixed and the excitation wavelength is scanned, the resulting spectrum is termed an excitation spectrum (Figure 1). The fluorescence spectrum is plotted as relative intensity or counted photons of fluorescence vs. wavelength. The appearance of a fluorescence spectrum is much like a UV–Vis–NIR absorption spectrum. In fact, the shape or contour of an excitation spectrum often is identical to that of the corresponding absorption spectrum for an organic dye in solution over the same wavelength range.
Polyatomic fluorophores in condensed media (e.g., solutions, thin films, and solids at room temperature) exist in ground or excited electronic states in a broad distribution of vibrational energy levels and cause homogeneous broadening of excitation or emission spectra, respectively. A microenvironment or shell also surrounds each fluorophore in condensed media, and differences in the structures of these shells among individual fluorophores cause inhomogeneous broadening. These two types of broadening cause fluorescence spectra to be broader than some other types of spectra (e.g., mid-infrared or Raman spectra). The typical width of a fluorescence band is between 10 nm and 100 nm. Once it is electronically excited, a polyatomic fluorophore experiences vibrational relaxation before emitting a photon, causing a red shift or Stokes shift of the fluorescence spectrum relative to the wavelength at which it was excited.
Few naturally occurring biological compounds fluoresce strongly.
A right-angle or 0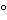 /90 geometry often is used to measure dilute solutions and other transparent samples. In such cases, the excitation beam is normal to the sample, and fluorescence is detected at a 90 angle relative to the beam (Figure 2a). A front-face geometry is used to measure optically dense samples where the excitation beam is incident on the sample at <90 and the fluorescence is collected at an angle
/90 geometry often is used to measure dilute solutions and other transparent samples. In such cases, the excitation beam is normal to the sample, and fluorescence is detected at a 90 angle relative to the beam (Figure 2a). A front-face geometry is used to measure optically dense samples where the excitation beam is incident on the sample at <90 and the fluorescence is collected at an angle 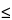 90 (Figure 2b). The epifluorescence geometry is a special case of the front-face geometry that often is used in fluorescence microscopy and optical fiber–based fluorometers. In epifluorescence geometry the excitation beam and collected fluorescence are both normal to and are on the same side of the sample, i.e., a 0/0 geometry. A 0/180 transmitting geometry often is used in microscopy (Figure 2c).
90 (Figure 2b). The epifluorescence geometry is a special case of the front-face geometry that often is used in fluorescence microscopy and optical fiber–based fluorometers. In epifluorescence geometry the excitation beam and collected fluorescence are both normal to and are on the same side of the sample, i.e., a 0/0 geometry. A 0/180 transmitting geometry often is used in microscopy (Figure 2c).
+'&img=../../images/v38332/m3207-f2-pf345.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 2. Schematic of the excitation beam (EX) and detected emission (EM) orientations for (a) 0/90 right-angle transmitting, (b) front-face, and (c) 0/180 transmitting geometries.
The number of chemical assays and screening methods using fluorescence detection continues to increase rapidly and has resulted in a corresponding increase in the need for standardization of fluorescence measurements. Only a few standard methods and reference materials have been well established and are readily available at present for the characterization of fluorescence measuring systems. National metrology institutes and international standards organizations are working to provide new fluorescence standard materials and methods in the near future. This general chapter briefly discusses the major issues that should be considered by users of fluorescence instruments who aim to achieve high-quality measurements. Standard methods and materials are also described where appropriate. A few guidelines and recommendations have appeared, but this general chapter aims to be most useful to nonexpert users of fluorescence spectrometers.
INSTRUMENTATION
All modern fluorescence measurements involve irradiating the sample with the beam from a suitable light source, selecting the excitation wavelength, collecting the resulting fluorescence, rejecting the Rayleigh-scattered light, selecting the emission wavelength, and detecting the fluorescence signal. The following functions will be discussed individually, along with the equipment used to achieve these functions in commercial instruments:
-
excitation light source
-
excitation wavelength selector
-
sampling device
-
emission wavelength selector
-
detector.
Excitation Light Source
A variety of lamps, lasers, and light-emitting diodes (LEDs) are used as excitation sources. Continuous and pulsed versions of these sources are used for steady-state and time-resolved instruments, respectively. Xenon lamps are the most commonly used because of their relatively high intensity and broad wavelength range (UV to NIR). Lasers are the highest-intensity sources and are used in applications where short collection times and small amounts of sample are required, e.g., for flow cytometry or microarrays.
Excitation Wavelength Selector
The intensity of scattered light at the excitation wavelength (i.e., Rayleigh scattering) can be comparable to or greater than that of the fluorescence at the sample. Therefore, the excitation wavelength profile should not overlap the emission wavelength region being detected. This is achieved for lamps by using an excitation wavelength selector (e.g., a filter or a monochromator with a known peak transmission wavelength and bandwidth) between the lamp and sample. The inherent bandwidth of the radiation from a laser or an LED often is narrow enough that an excitation wavelength selector is not necessary. This selector also enables fluorescence excitation spectra to be resolved.
Sampling Device
The sampling device includes all optics and other equipment needed to deliver the excitation beam to the sample, collect the emission from the sample, and hold the sample in place. Sample formats include cuvettes, microwell plates, microarrays, microscope slides, and flow systems and may be accompanied by a variety of optical delivery and collection systems, including conventional transmitting, front-face, and epifluorescence systems and fiber optic–based probes.
Emission Wavelength Selector
As with the selector for excitation, the emission wavelength selector helps to ensure that the emission wavelength region being detected does not overlap with the excitation wavelength profile. This approach enables individual fluorescence bands to be detected when multiple bands are present and allows fluorescence emission spectra to be resolved. Emission wavelength selectors also are important for the rejection of stray light. Filters, monochromators, and grating polychromators often are used for emission wavelength selection.
Detector
For the detection of emitted light, a photomultiplier tube (PMT) or a charge-coupled device (CCD) array is placed after the emission wavelength selector. The detection of the excitation beam in order to monitor its intensity commonly is done by a quantum counter detector or a photodiode placed before the sample and to which a small fraction of the excitation beam is split off from the rest.
FACTORS THAT AFFECT QUANTITATION
Instrument-Based Factors
Measurements on a fluorescence instrument require that instrument parameters such as wavelengths, bandwidths, and detector gain be set. All of these parameters can be set with varying degrees of repeatability and accuracy, depending on the instrument used. These factors can introduce measurement uncertainty or bias that is particularly significant when measured values are compared between instruments. For instance, the measured peak positions of the emission bands of two analytes may differ between instruments because of a wavelength bias. A corresponding bias between instruments could be introduced in the results of an assay that depends on the ratio of the fluorescence intensities at the two specified emission wavelengths.
The intensity of the excitation beam can change significantly with excitation wavelength or with time because of the wavelength dependence of the intensity of the light source and the transmittance of the excitation wavelength selector or the time dependence of the light source intensity. Thus analysts should monitor the excitation beam intensity and correct the measured fluorescence intensity for these fluctuations. This monitoring can be particularly important when excitation spectra are collected because the excitation intensity often has sharp peaks and valleys when lamp sources such as a xenon (Xe) lamp are used.
The responsivity of a detection system is not linear with intensity at all intensities, so analysts should know the linear intensity range of the detection system used. The linear range for most detection systems ranges from its limit of detection up to a threshold intensity above which the responsivity becomes increasingly nonlinear with increasing intensity. Analysts should establish the linear range of the fluorescence detection system before they attempt to calibrate the responsivity of the detection system.
The responsivity of the detection system also is a result of the wavelength dependence of the transmittance of the emission wavelength selector and the responsivity of the detector. These factors can affect the shape of a measured emission spectrum.
The diffraction efficiency of gratings and the responsivity of detectors often depend on polarization. Changes to instrumental polarization settings can result in changes in the observed excitation intensity and the responsivity of the detection system. Even when polarizers are not used within the instrument, the excitation beam may be polarized by the optical system itself and may affect the responsivity of the detection system, and is instrument dependent. In addition, emission polarization effects not only can cause intensity differences but also can change spectral correction factors.
The passing of multiple wavelengths by a diffraction grating can introduce unexpected sharp peaks into a fluorescence spectrum. So that incident light is diffracted at a desired wavelength, a grating equation is used to set the angle of the grating with respect to incident light:
m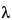 = d(sin
= d(sin 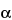 + sin
+ sin 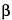 ), m = 0, 1, 2, ...
), m = 0, 1, 2, ...
m = diffraction order
d = groove spacing on the grating
The value of m, not , is fixed, where m is an integer termed the diffraction order. Therefore, the grating equation can be satisfied by more than one wavelength for a single grating position. For instance, if a grating in an emission monochromator is set to pass 500-nm light at first order, it also will pass 250-nm light at second order. As a result, the scattered light from a 250-nm excitation beam will be detected as a peak at an emission wavelength of 500 nm unless a suitable optical filter is inserted in the beam.
Sample-Based Factors
The fluorescence intensity of optically dense samples (e.g., absorbance A >0.05 at a path length of 1 cm) does not increase linearly with concentration because of significant absorption of the excitation beam and/or emission (reabsorption) by the sample. These inner filter effects also can greatly reduce the amount of fluorescence that reaches the detector, especially when a right-angle transmitting geometry is used. The fluorescence intensity can become strongly dependent on sample position and optical geometry. At even higher concentrations, aggregation of fluorophores often occurs, causing the shape of the fluorescence spectrum to be different from that of a dilute sample and also causing nonlinear concentration behavior.
The fluorescence intensity of a sample may decrease with time of exposure to light because of photobleaching and photodegradation. This is particularly true of most organic dyes, which are the most widely used fluorescent probes. Analysts should limit the time that such samples are exposed to light in order to obtain reproducible fluorescence intensities and in some cases even reproducible spectral shapes.
The fluorescence intensity of fluorophores is temperature dependent. Typically, the rates of fluorescence quenching processes, such as collisional quenching in solutions, increase with temperature and cause a decrease in fluorescence intensity. Temperature coefficients for fluorescence intensity for particular fluorophores can be used to correct for this temperature dependence.
The absorbance and consequently the intensity of fluorescence from a sample depend on the orientation of the sample's absorption transition dipole with respect to the polarization of the excitation light. The polarization of fluorescence is parallel to the direction of polarization of the fluorescent species' emission transition dipole. Fluorescence polarization is parallel to the orientation of the fluorescent species' emission transition dipole. Fluorescence anisotropy (r) is used to describe the extent of polarization of emission and is defined by:
r = (I|| 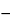 I
I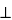 )/(I|| + 2I)
)/(I|| + 2I)
I|| = observed fluorescence intensity when the fluorometer's emission polarizer is oriented parallel to the direction of the polarized excitation
I
A sample whose fluorophores are oriented nonrandomly and have a rotational period that is long compared to their fluorescence lifetime will emit anisotropic fluorescence. The spectral shape and intensity of such fluorescence depend on the viewing angle and the instrument's polarization factors.
A fluorophore's fluorescence intensity and peak position, and sometimes even its spectral shape, often depend on the environment, including changes caused by the solvent used, the solution's pH, or the species to which the fluorophore is bound.
A Raman signal can introduce peaks into the fluorescence spectrum. The Raman peaks of the sample's solvent or matrix are those most commonly encountered. For instance, the Raman peak of water, which is found red-shifted
CALIBRATION OF FLUORESCENCE INSTRUMENTS
Two types of fluorescence instrument calibrations are used. The first and most commonly used is analyte specific and determines the relationship between the response of the instrument (fluorescence intensity) and the concentration or amount of a specific analyte. The second is analyte independent and is intended for spectral instruments. In this case, the wavelength accuracy for emission and excitation and the spectral responsivity of the detection system are calibrated across the entire or a continuous part of the wavelength range of the instrument.
Analyte Concentration—Calibration Curves
Calibration curves of instrument response (fluorescence intensity) vs. analyte concentration are determined using reference materials that contain the analyte of interest. For instance, the fluorescence intensities of a set of solutions at different, known analyte concentrations that cover a desirable concentration range can be measured and plotted vs. concentration. The plot then is fitted to a polynomial, typically a straight line. The resulting calibration curve is both analyte and instrument specific and can be used to determine analyte concentrations of unknown samples. This type of calibration may not be accurate when the microenvironment surrounding the fluorophore is different in the reference and unknown samples. In addition, users must ensure that the fluorescence intensities of samples are reproducible and do not decrease over the time when they are being excited and measured because the organic dyes typically used may be prone to photobleaching.
In some cases, calibration samples at known concentrations and prepared from appropriate reference materials may not be available. Firstly, organic dyes that are used as fluorescent probes often are not commercially available at a known high purity that enables production of reference solutions. Secondly, in complex systems where fluorophores are bound to large molecules, cells, or microbeads, the concentration of bound fluorophores in a solution or suspension may be difficult to determine. In the latter case, the molecules of equivalent soluble fluorophore (MESF) scale has been proposed as an alternative way to use calibration curves to quantify fluorescence intensity for a particular analyte.
Emission Wavelength and Spectral Slit Width
A variety of reference standards have been proposed for use in the determination of emission wavelength accuracy, including atomic lamps and inorganic and organic fluorophores. The most widely used and best characterized of these are low-pressure atomic lamps, commonly termed pen lamps. In this case, the type of pen lamp (e.g., Hg,
The spectral slit width accuracy of the emission wavelength selector can be confirmed by measuring the spectral bandwidth, taken to be the full width at half the peak maximum, of a single line of a pen lamp. For fluorescence spectrometers with both excitation and emission monochromators, an alternative method can be used when one monochromator is scanned over the position of the other.
Excitation Wavelength and Spectral Slit Width
Many of the reference samples that are used for determining emission wavelength accuracy also can be used for excitation wavelength accuracy. For instance, a pen lamp can be placed at the excitation light source position so that the resulting spectrum is detected after the excitation wavelength selector using the instrument's reference detector. However, in this case, a relatively weak signal may limit the number of useful atomic lines, and therefore alignment of the pen lamp is more critical in this instance than for the emission wavelength accuracy determination.
Once the accuracy of the emission wavelength selector has been determined, use a diffuse scatterer, e.g., a scattering solution or a diffuse reflector, at the sample to scatter a fraction of the excitation beam into the detection system to determine excitation wavelength accuracy. One wavelength selector is set at a fixed wavelength and the other is tuned over the same wavelength to obtain a spectrum. The wavelength bias between the two wavelength selectors is equal to the difference between the set wavelength position and the observed peak position of the collected spectrum. This method can be used at any wavelength, unlike many other methods that depend on a limited number of set excitation wavelengths determined by the reference material chosen. Methods similar to those used for spectral slit width accuracy of the emission wavelength selector also can be used to determine the spectral slit width accuracy of the excitation wavelength selector.
Linearity of the Detection System
Several approaches are available to determine the detection system's linear intensity range. They can be separated into three types, based on the tools used to vary the intensity of light that reaches the detector: (1) double aperture, (2) optical filters and/or polarizers, and (3) fluorophore concentrations. The double-aperture method is the best established and probably is the most accurate when done correctly, but it also is the most difficult to perform. A variety of methods using optical filters, polarizers, or a combination of the two have been reported. These methods require high-quality, often costly, components and some user expertise. The third method is the most popular and is easiest. It uses a set of solutions obtained by serial dilution of a fluorescent stock solution that is similar to one used for obtaining calibration curves for analyte concentration, as described earlier. In this case, analysts use solutions with low concentration (A <0.05 at 1-cm path length), but fluorophore adsorption to cuvette walls may affect measurements at very low concentrations. Users must ensure that the fluorescence intensities of samples are reproducible and do not decrease over the time that they are being excited and measured because the organic dyes typically used may be prone to photobleaching.
Signal Level (Relative Emission)
Calibration of the relative responsivity of the emission detection system with emission wavelength, also referred to as spectral correction of emission, is necessary for successful quantification when intensity ratios at different emission wavelengths are compared or when the true shape or peak maximum position of an emission spectrum must be known. Such a calibration is required because the relative spectral responsivity of a detection system can change significantly over its wavelength range (Figure 3). Analysts should know the degree of photometric precision required for successful quantitation. The linear range of the detection system is determined before this calibration is performed, so that appropriate steps are taken (e.g., the use of attenuators) to ensure that all intensities measured during this calibration are within the linear range. When one uses an emission polarizer, the spectral correction for emission depends on the polarizer setting.
Two methods are preferred for calibrating photometric responsivity: one (Method A) uses light from a calibrated source (CS), and the other (Method B) uses certified reference materials (CRMs). Both give results that are traceable to national metrology institutes. A calibrated tungsten white light source is used most commonly for Method A and covers the wavelength range from about 350 nm into the NIR. Standard reference materials from the U.S. National Institute of Standards and Technology and CRMs from the German Federal Institute for Materials Research and Testing currently are available for use in Method B. Corrected emission spectra of some commonly used dyes also have been reported in the literature. Method A is more difficult to implement than Method B and requires periodic recertification of the CS. A third method, Method C, uses a calibrated detector and a calibrated diffuse reflector. This method typically has larger uncertainties than Method A and Method B but is recommended in UV and NIR wavelength regions that are not covered by the other two methods.
+'&img=../../images/v38332/m3207-f3-pf345.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 3. Example of the relative spectral responsivity of an emission detection system (PMT-based grating monochromator) for which a correction must be applied to a measured emission spectrum to obtain the true spectral shape (relative intensities).
method a
In Method A, analysts direct CS light into the emission detection system by placing the CS at the sample position. If the CS is too large to be placed at the sample position, analysts can place a calibrated diffuse reflector (CR) at the sample position to reflect the light from the CS into the emission detection system. The emission wavelength selector is scanned over the emission region of interest using the same instrument settings as used with the sample, and the signal channel output (S¢¢) is collected. The known radiance of the CS incident on the detection system (L) can be used to calculate the relative correction factor (CCS) so that:
CCS = L/S¢¢
CCS = relative correction factor
L = radiance of the CS incident on the detection system
S¢¢ = signal channel output
The corrected emission intensity is equal to the product of the signal output of the sample and CCS.
method b
In Method B, analysts place the fluorescence standard at the sample position. Its spectrum is collected and is compared to the certified spectrum according to the instructions given on the accompanying certificate, which yields spectral correction factors for the instrument.
method c
Method C involves two steps: Step 1 uses a calibrated detector (CD) at the sample position to measure the flux of the excitation beam as a function of excitation wavelength. Step 2 uses a CR to reflect a known fraction of the flux of the excitation beam into the detection system. This is done by placing the CD at the sample position at a 45 angle, assuming a 0/90 instrument geometry, and synchronously scanning both the excitation and emission wavelength selectors over the emission region of interest while collecting both the signal output and the reference output. This method allows analysts to calculate the relative correction factor. This method has larger uncertainties than those for Method A or Method B and typically is more difficult to implement.
Reference Signal Level (Relative Excitation)
Calibration of the excitation intensity with excitation wavelength is necessary for successful quantitation when analysts compare intensity ratios at different excitation wavelengths or when analysts must know the true shape or peak maximum position of an excitation spectrum. Such a calibration is necessary because the relative spectral flux of an excitation beam at the sample can change extensively over its wavelength range (see Figure 4). The neglect of excitation intensity correction factors can cause even greater errors than neglect of emission correction factors. Fortunately, many fluorescence instruments have a built-in reference detection system to monitor the intensity of the excitation beam. This monitoring usually is done using a photodiode, PMT, CCD, or a quantum counter detector to measure a fraction of the excitation beam that is split off from the rest of the beam. The collected reference signal can be used to correct the fluorescence signal for fluctuations caused by changes in the excitation beam's intensity. Reference detectors often are not calibrated with excitation wavelength, which introduces errors that can be particularly large over longer excitation wavelength ranges (e.g., >50 nm) or in a wavelength region where the excitation intensity changes rapidly with excitation wavelength (e.g., the UV range). When an excitation polarizer is used, the spectral correction for excitation intensity depends on the polarizer setting.
+'&img=../../images/v38332/m3207-f4-pf345.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 4. Example of the relative flux of an excitation beam (Xe lamp grating monochromator) for which a correction must be applied to a measured excitation spectrum in order to obtain its true spectral shape.
When a reference detector is not built into an instrument, a spectral correction for the reference channel or an independent spectral correction of excitation intensity is required. Three methods can be used to determine the spectral correction of excitation intensity: a CD (Method 1); a calibrated diffuse reflector (Method 2); or a quantum counter (Method 3). The latter two methods use the instrument's fluorescence detection system as a detector.
For Methods 1 and 2, the detector and diffuse reflector are calibrated for responsivity and reflectance, respectively, as a function of wavelength. For Method 2, excitation and emission wavelength selectors are scanned synchronously, and the spectral correction for the emission channel [see Signal Level (Relative Emission)] must be applied to the measured intensities. Method 3 should be used only in the quantum counter's effective wavelength range where a wavelength-independent response can be achieved. Method 1 using a CD has fewer caveats than do the other two methods. A CD is placed in the sample position, and the output is measured as a function of emission wavelength by scanning the excitation wavelength selector over the excitation region of interest using the same instrument settings as those used with the sample. The known responsivity of the CD is used to calculate the flux of the excitation beam. If the instrument's reference detector is used to measure the intensity of the excitation beam simultaneously with the CD, then the correction factor for the responsivity of the reference detector also can be calculated.
Intensity and Sensitivity
The absolute value of the fluorescence signal measured by the detection system depends not only on the sample itself but also on the excitation intensity of the sample and the optical geometry of the instrument. Therefore, determination of instrument-independent fluorescence intensity of any sample or the absolute responsivity of any detection system in terms of the intensity of the sample or measured by the detector, respectively, relative to the excitation intensity can be difficult.
The most accurate way to calibrate an instrument for absolute intensity is to use conventional standards-based methods such as those that employ a calibrated light source or a calibrated detector in combination with a calibrated reflector. These methods require user skill and knowledge. Also, the (typically annual) certification and recertification are expensive. In addition, these standards tend to be bulky and are not compatible with many instruments. Thus, most researchers use simpler alternative standards and methods.
One approach is to correlate fluorescence signals to analyte concentrations using calibration curves or MESF units (see Analyte Concentration—Calibration Curves). Another approach is to measure the intensity of a standard sample that can be expected to always give the same fluorescence intensity under the same conditions.
Organic dyes, such as those used as fluorescent probes, generally are not good choices for intensity standards because of issues with photobleaching, stability, and reproducible concentration. If organic dyes are used, then those with known high purity and known shelf life, such as those produced by national metrology institutes, are recommended for single use (i.e., analysts should use a fresh solution for every measurement).
A better alternative is to use fluorescent samples that are stable over time even when exposed to light. For example, fluorescent, inorganic glasses with well-characterized photostability and spectral properties and long shelf lives are commercially available. Such materials can be used for determining a quasi-absolute intensity scale by measuring fluorescent signal at fixed wavelength values within their recommended range using specified experimental parameters such as bandwidths, excitation intensities, and temperatures.
The sensitivity of a fluorescence instrument is determined by measuring the signal-to-noise ratio of the fluorescence signal of intensity standards. The Raman line of water often is used to measure sensitivity in a similar way, but the Raman signal typically is strong enough only to be useful in the UV region. Analysts can use organic dye solutions to measure instrument sensitivity or limits of detection with caveats that are identical to those that apply when the solutions are used as intensity standards.
The methods outlined here yield a quasi-absolute intensity scale that should be instrument independent for instruments with similar optical geometries, designs, and settings. Results of these measurements enable comparison of the sensitivity of different fluorescence instruments, but these comparisons should be approached with caution because of the relatively large and difficult-to-quantify uncertainties involved.
PROCEDURE VALIDATION
Validation of an analytical procedure using fluorescence demonstrates that the result is valid within a specified, acceptable uncertainty budget. Instrument qualification, which also may involve instrument calibration, usually is part of the process, and analysts also must consider sample-related errors (see Sample-based Factors). These can arise from concentration, anisotropy, photostability, and shape of the sample, in combination with effects of the instrument's optical geometry. All suspected errors should be quantified and combined to give a total estimated error that must be less than the method-specific, acceptable limit.
APPENDIX: DEFINITIONS
Absorptivity (a):
A = absorbance
b = path length (cm)
c = concentration (mg/mL)
A/bc
A = absorbance
b = path length (cm)
c = concentration (mg/mL)
Also referred to as specific absorption coefficient by the International Union of Pure and Applied Chemistry.
Absorption coefficient ():
I = transmitted intensity
I0 = incident intensity
e = base of natural logarithm
a = absorptivity
b = path length of the beam through the sample
I/I0 = e–ab
I = transmitted intensity
I0 = incident intensity
e = base of natural logarithm
a = absorptivity
b = path length of the beam through the sample
Note that transmittance T = I/I0 and absorbance A = log T.
Beer–Lambert law (or Beer's law or Beer–Lambert–Bouguer law):
 = molar absorptivity
= molar absorptivity
c = solute concentration (M/L)
b = path length (cm)
A = cb
c = solute concentration (M/L)
b = path length (cm)
Calibrated detector (CD):
Calibrated light source (CS):
Calibrated diffuse reflector (CR):
Certified reference material (CRM):
Diffuse scatterer:
Fluorescence anisotropy (r):
I|| = observed fluorescence intensity when the fluorometer's emission polarizer is oriented parallel to the direction of the polarized excitation
I = observed fluorescence intensity when the fluorometer's emission polarizer is oriented perpendicular to the direction of the polarized excitation
r = (I|| I)/(I|| + 2I)
I|| = observed fluorescence intensity when the fluorometer's emission polarizer is oriented parallel to the direction of the polarized excitation
I
Fluorescence band:
Fluorescence lifetime:1
Fluorescence quantum efficiency:
Fluorescence quantum yield (F):
Flux (or radiant flux):
Grating equation:
m = diffraction order
d = groove spacing on the grating
= angle of the incident wavefront relative to the grating normal
= angle of the diffracted wavefront relative to the grating normal
m = d(sin + sin )
m = diffraction order
d = groove spacing on the grating
Inner filter effects:
Intensity:
Lambertian reflector:
Limit of detection (LOD):
Noise level:
Photobleaching:
Quantum counter:
Quasi-absolute fluorescence intensity scale:
Raman scattering:
Rayleigh scattering:
Responsivity (spectral):
Sensitivity:
Spectral bandwidth (or spectral bandpass or resolution):
Spectral slit width:
Transition dipole moment:
 USP38
USP38
1
Boens N, Qin W, Basaric N, et al., Fluorescence lifetime standards for time and frequency domain fluorescence spectroscopy. Anal Chem. 2007;79(5):2137–2149.