



Add the following:
INTRODUCTION
Parenteral products are designed and manufactured to minimize particulate matter, which is differentiated into two broad categories: visible and subvisible. The absolute limit of visibility, or detectability, depends on the test conditions and the nature of the particulate matter. This general information chapter covers subvisible particles in the range of 2–100 µm. Particles larger than 100 µm are generally considered visible particles (see Visible Particulates in Injections  790
790 ).
).
Foreign and particulate matter are undesirable in the final product. These undesired particles arise from two sources: extrinsic or foreign matter and intrinsic or product-related matter. Extrinsic matter cannot be associated with product or process. Intrinsic particles may be a byproduct of the assembly of the product or result from change over time. A third category, inherent matter, describes a physical state or particles that are an expected attribute of the product. The aim of this chapter is to provide additional information to support Subvisible Particulate Matter in Therapeutic Protein Injections 787, which has limits for 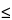 10-µm and 25-µm particle content. Because the monitoring of the sub-10-µm population may be an important product quality parameter, collection of data in the 2–10-µm range (e.g. 2–5 µm and 5–10 µm) is recommended. This chapter will focus on enumeration, characterization, and when possible, identification of inherent particles, distinguishing them from extrinsic and intrinsic particles. The chapter does not cover formulations that are suspensions or emulsions, or those that contain adjuvants or similar intended particle components. However, foreign and particulate matter should be minimized in the final drug product, and the strategy and methods utilized are ultimately determined by the manufacturer.
10-µm and 25-µm particle content. Because the monitoring of the sub-10-µm population may be an important product quality parameter, collection of data in the 2–10-µm range (e.g. 2–5 µm and 5–10 µm) is recommended. This chapter will focus on enumeration, characterization, and when possible, identification of inherent particles, distinguishing them from extrinsic and intrinsic particles. The chapter does not cover formulations that are suspensions or emulsions, or those that contain adjuvants or similar intended particle components. However, foreign and particulate matter should be minimized in the final drug product, and the strategy and methods utilized are ultimately determined by the manufacturer.
Extrinsic particles: Materials that are not part of the formulation, package, or assembly process, but rather are foreign and unexpected. Examples of extrinsic particles include materials from biological sources (e.g., fibers, insect parts, pollens, and vegetative matter), from process and building materials (e.g., cellulose, lint, minerals, glass, plastics, rubber, metal, and paint), and from personnel (e.g., epithelial cells, clothing fragments, and hairs).
Intrinsic particles: Materials occurring in the final product that arise from sources within the formulation ingredients, assembly process, or packaging. Their association with the production process and/or package-related sources can be indicative of systemic problems. For example, silicone oil is an important manufacturing and product component that may affect particle counts and, in excess, is considered intrinsic. The incidence of silicone oil-related particles may correlate with processing sequences or formulation component sources. Detection, identification, and measurement of silicone oil-related particles are discussed further in another section of this chapter. Intrinsic particles can be similar in some ways to extrinsic particles such as rubber, metal, plastic, and glass, but may arise because of insufficient cleaning of processing equipment. Intrinsic particles can also arise from changes in the product over time. These changes may be related to ionic or organic extracts, instability of the active pharmaceutical ingredient (API), formulation degradation, or product–package interaction. Intrinsic particles may promote protein, and one should be aware of this possibility.
Inherent particles: Materials that are expected from the drug substance and other formulation components, and thus represent a potentially acceptable characteristic of the product. In the context of therapeutic protein injections, the primary source of inherent particles is proteinaceous aggregates formed either solely by interactions of the protein with itself or in combination with other formulation ingredients. The presence of proteinaceous particles should be assessed for process consistency and product quality. Identification and characterization of the aggregates are critical for distinguishing them from extrinsic and intrinsic particles. The tools and techniques discussed in this chapter are applicable to all sources but are especially useful for the characterization of protein aggregates.
Heterogeneous particles consist of more than one chemical entity. They are classified as either extrinsic or intrinsic based on the nonproteinaceous component that presents the greatest risk.
Historically, test methods contained in Particulate Matter in Injections 788 were developed for detection and control of extrinsic and intrinsic particle content and rely on light obscuration (LO) and/or microscopic determination of particles that are between 10- and 25-µm thresholds. These sizes were selected to allow consistent measurement of subvisible particle content and allow determination of product acceptability from the perspective of patient safety and process consistency. The membrane microscopic method was developed first and was used only for large-volume (>100 mL) products. Later, improvements in the microscopic method and development of an automated light extinction (obscuration) method provided an efficient and robust means of tracking particulate content in a variety of products with different fill volumes. Evolution of analytical application is typical of the compendial standard-setting process, where methods are updated to reflect technological advances, patient safety considerations, and regulatory requirements.
Since 788 became official, the varieties of dosage forms and the numbers of therapeutic protein products have increased markedly. The LO method described in 788 has some technical limitations when used for analyzing certain particle types (e.g., those that have low contrast in the product medium and/or may change shape/size during analysis, as is typical for inherent particles in therapeutic protein injections). Other 788 limitations for therapeutic protein products relate to sample handling and low product volumes. These considerations have led to the development of Subvisible Particulate Matter in Therapeutic Protein Injections 787.
OBJECTIVE
This chapter was written to describe strategies for identifying and characterizing extrinsic and intrinsic particle populations, compared with inherent proteinaceous particle population(s), in therapeutic protein injections. Information about specific methods that can be used for these purposes, as well as their advantages and limitations, is discussed; the information applies to therapeutic protein injection products and their dilution or infusion solutions. The overarching goal is to provide comprehensive guidance on use of a broad array of analytical methods, one or more of which may be used beyond the methods described in 787 and 788. This is expected to achieve enhanced or orthogonal characterization of therapeutic protein products during development and support, root cause analysis for nonconformance investigations, stability studies, and others. This includes methods that allow assessment of a variety of characteristics of the inherent protein aggregates including morphology, conformation, reversibility/dissociation, and covalent modification. Guidance on sample handling and preparation is discussed in general terms.
BACKGROUND
Extrinsic and intrinsic particles should be minimized in all parenteral products. However, protein-related particles may be inherent to therapeutic protein products. Inherent particles should be understood and controlled. These inherent particles are known and expected, arising from the association of protein molecules that can be present in a continuum of sizes, ranging from nanometers (dimers) to hundreds of micrometers (multimers and visible particles). The distribution of particle size is affected by amino acid sequence, solution conditions, sample handling history, and other factors. The specific phenomenon of protein association that gives rise to particles is called aggregation. The terms “proteinaceous particles” and “aggregates” are used interchangeably in this chapter.
Because multiple potential sources of particles exist, it is important to identify the particles and determine whether they are extrinsic, intrinsic, or inherent. Once this has been accomplished, it is possible to develop and apply appropriate control strategies. If deviations occur, particle identity will guide the root cause analysis, risk assessment, corrective actions, and control strategy.
Protein aggregates inherent in therapeutic protein products may consist of a heterogeneous population, and therefore size alone does not adequately describe them. When aggregate populations are being compared across products, labs, etc., it is important to consider other characteristics such as morphology, the ability to dissociate, the protein conformations (or higher-order structures) within the aggregates, and the chemical modifications present (1). These are summarized in Table 1 [adapted from (1)]. It is important to realize that the results obtained depend on the technique and methodology used, and this information must be included in any description of protein aggregate characterization. The tools available for aggregate characterization are described later in this chapter. The tools used and the characteristics analyzed should be thoughtfully chosen based on the protein being studied. A more detailed description of attributes that should be considered during aggregate characterization follows.
Size is the particle attribute that has been monitored historically and remains a primary descriptor. When measurements are reported, they should include the size range measured and technique used. Thus, subvisible particles could also be described as aggregates/particles between 1 and 100 µm based on: the equivalent circular diameter, determined using LO; the longest chord, using light microscopy; the equivalent spherical volume, using electrical zone sensing; or other relevant dimensions obtained using other available technologies. The sample preparation, determination method, and algorithms applied are all important factors to include when presenting the results.
The ability of any aggregate species to dissociate (i.e., revert to the monomeric state) and the conditions required to accomplish this are also important characteristics to consider. Sample preparation could affect the results obtained if the aggregates are dissociable; therefore, understanding this aspect of the aggregates is critical when selecting techniques for counting, measuring, and characterizing them. The dissociability of protein aggregates can be assessed by diluting the aggregates into the formulation buffer or other solutions and then re-analyzing them with the original technique. This can also include simply returning the sample to the original conditions and assessing how much of the aggregate has dissociated. Ability to dissociate is thus a characteristic of protein aggregates that can inform sample handling and preparation (e.g., dilution conditions) for particle counting. The ability to dissociate may also apply to intrinsic and, to a lesser extent, extrinsic particles species.
The structure and conformation of protein molecules in the aggregates can be investigated by using several biophysical techniques. The findings may help in investigations and troubleshooting. It is also useful to know about the presence of chemical modifications such as oxidation, deamidation, cross-links, or fragments in the aggregate. Conformation of protein molecules within protein aggregates, as well as their state of covalent modification, may help explain the potential biological impact of the inherent particle.
Morphology of the particles can serve as another descriptor and also help identify their source. More specifically, characterization of morphology may allow one to distinguish between particles that are inherent to the therapeutic protein product and those that are extrinsic or intrinsic. Proteinaceous as well as many intrinsic and extrinsic particles are generally of irregular shape, whereas silicone oil and air bubbles tend to be spherical. The morphology of protein aggregates also constitutes important data that can facilitate comparisons across studies and over time.
Table 1 provides an overview of protein aggregate characteristics to consider, which can be used to classify them.
Table 1. Description of Categories of Protein Aggregates (1)
| Category |
Classification |
|---|---|
| Size |
<100 nm (nanometer) 100–1000 nm (submicrometer) 1–100 µm (subvisible) >100 µm (visible) |
| Dissociability |
Reversible (revert to initial state when returned to original conditions) Irreversible (do not dissociate under conditions tested) Dissociable (dissociate under specific conditions tested, e.g., dilution, changes in buffer, etc.) Dissociable under physiological conditions |
| Reversibility |
Reversible Irreversible Dissociable Dissociable under physiological conditions Dissociable under defined (list) conditions |
| Secondary/tertiary structure |
Native Partially unfolded Unfolded Inherently disordered Ordered (e.g., amyloid) |
| Covalent modification |
Cross-linked (reducible and nonreducible) Intramolecular modification Oxidation Deamidation Fragmentation No modification |
| Morphology |
Number of monomeric subunits Aspect ratio Surface roughness Internal morphology Optical properties Heterogeneous (e.g., protein–silicone oil) |
Silicone oil: A standard lubricant in pharmaceutical products and packaging. In excess, it may contribute to elevated particle counts in products. Assessing the effect of silicone oil on particle counts in therapeutic protein injections, where relevant, is critical to the overall particle control strategy. In parenteral manufacturing, silicone oil is used primarily to facilitate packaging component handling, alleviate adherence of rubber parts, allow free stopper flow in the bowl/hopper, and promote tracking onto the waiting, filled container. Silicone oil is also used as a lubricant to decrease glide force and allow plunger stopper movement in glass, pre-filled syringe presentations. For parenteral container preparation, controlled stages of washing, rinsing, siliconization, and sterilization are routine. The tumbling of closures (e.g., vial stoppers) with a measured amount of silicone has been a routine process for uniformly coating surfaces before use. Application of a known amount of an accepted lubricant, rendered sterile, is common.
Even when applied judiciously, silicone oil can migrate from the product-contact surface to the fill-over time during product transport, storage, and use. Silicone oil droplets can contribute significantly to the subvisible particle counts of the product, specifically, particles in the <10-µm range in the case of therapeutic protein injections. The particle count depends on the amount of silicone oil, and in some cases small amounts of silicone oil can have a significant effect on particle counts (2).
Within therapeutic protein products, protein may become adsorbed to silicone oil droplets. Agitation resuspends and rearranges the oily matrix into different particle forms and sizes. The size range of the silicone oil droplets in protein formulations may be similar to inherent protein particles, but the morphology and optical properties of the two substances differ. Silicone oil in aqueous solution is typically spherical, with a higher refractive index than that of protein aggregates and a regular decrease in contrast from the outside to the center of the particle. This is unlike protein aggregates, which are typically amorphous and translucent and have a refractive index very similar to the protein monomer background of the drug formulation. These distinctions can be used to develop algorithms for differentiating silicone oil particles from protein particles. It is also possible to monitor changes in one population and distinguish them from changes in the other.
Because silicone oil serves a function in the product, particle counts arising from the oil are considered intrinsic and generally will vary over time. However, poorly controlled application processes can lead to excessive amounts of silicone oil. Careful design, control, and application of the silicone oil are recommended to obtain proper functionality with the minimum amount needed for the shelf life of the product.
PARTICLE STANDARDS
Analysis of particle size distribution depends on the instrument used and its method of calibration. Some available particle standards (count and size) are polystyrene latex beads, mono-disperse silica or polymethylmethacrylate (PMMA) beads, and polydisperse glass beads. All of these standards are spheres, with a refractive index and density that are distinctly different from those of therapeutic protein products. Because typical protein particles have irregular morphology and low optical contrast, there are currently no standards available to consistently mimic the morphology and parameters of protein particles.
Protein particles have a refractive index that is quite close to that of the matrix solution. This difference in refractive index is a critical parameter for detection of protein particles by methods that rely on light interaction to measure size distribution. It is important to establish standard reference materials that mimic the properties of protein particles (3). The development of standards that better resemble protein particles is currently an active area of research. These standards should serve as surrogates for the actual protein particles in that the surrogate's properties mimic those of actual proteins for the detection method of choice. This approach is necessary because standards using proteins are limited by the following factors: (a) storage and transport should be at temperatures close to 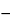 80
80 to achieve acceptable stability; (b) any non-ambient technique may affect the standard; and (c) protein particles are themselves quite variable, making it difficult to have a single protein that would match all applications.
to achieve acceptable stability; (b) any non-ambient technique may affect the standard; and (c) protein particles are themselves quite variable, making it difficult to have a single protein that would match all applications.
SUBVISIBLE PARTICLE MEASUREMENT AND CHARACTERIZATION TECHNOLOGIES
The following sections review the advantages, limitations, and appropriate uses of each methodology (see Table 2) that allow assessment of a variety of characteristics of all particles, especially for the inherent protein aggregates, including size and distribution, size and morphology, and characterization. The methodology selected will affect reported particle size because of instrumental measurement principles, mode of measurement, particle characteristics, preparation, and handling. It can also be affected by the algorithms applied to estimate size, volume, and count. Thus with any size measurement, the method used should be specified. Also, the units should be clearly stated, because size can be provided using various parameters, usually hydrodynamic diameter, equivalent circular diameter (ECD), longest chord, equivalent spherical diameter (ESC), Feret's diameter, or molecular weight. The list in Table 2 is not meant to be all inclusive.
Table 2. Methodologies Useful in Measuring Properties of
Subvisible Particles
Subvisible Particles
| Section I: Size and Distribution |
||
|---|---|---|
| Technique |
Principle of Operation |
Range |
| Light obscuration |
The size of the particle in the product fluid is determined by the amount of light that it blocks when passing between the source and the detector. |
1–300 µm |
| Electrical sensing zone (Coulter) |
The size of the particle in the product fluid or selected electrolyte is measured in terms of the change in resistance as the particle passes through a microchannel (orifice). |
0.4–1600 µm |
| Laser diffraction |
The size of the particles in product fluid or dilution is determined by measuring the angle of the scattered light. |
0.1–3500 µm |
| |
||
| Section II: Size and Morphology
|
||
| Technique
|
Principle of Operation
|
Range
|
| Light microscopy |
Photon imaging of substances directly in product fluids or mounts, or of isolated specimens on substrates |
0.3 µm to 1 mm |
| Flow imaging analysis |
Digital image capture of the particles' magnified image in streaming product fluid, revealing size, shape, and optical properties |
0.7–100 µm for size distribution; 4–100 µm for morphology |
| Electron microscopy (EM): Scanning EM, scanning transmission EM, and transmission EM |
Electron imaging of specimen isolates on substrates. High vacuum or near-ambient pressures is required. |
|
| |
||
| Section III: Characterization
|
||
| Technique
|
Principle of Operation
|
Range
|
| Fourier Transform Infrared (FTIR) microspectroscopy |
Photon imaging of isolated specimens on substrates |
10 µm to 1 mm |
| Dispersive-Raman microspectroscopy |
Photon imaging of isolated specimens on substrates, or in product fluids or fluid mounts |
0.5 µm to 1 mm |
| Electron microscopy (EM) with energy- dispersive X-ray spectrometry (EDS) |
X-ray photon emission from specimens energized by a focused electron beam |
1 µm to 1 mm for elemental composition |
| Electron microscopy (EM) with electron energy loss spectroscopy (EELS) |
Inelastic scattering from specimens energized by a focused e-beam; e-loss is characteristic of the source element; complementary to EDS |
|
| Time-of-Flight Secondary Ion Mass Spectrometer (TOF-SIMS) |
Identification of particles according to their mass spectra profile |
µm to near mm |
| Staining assay |
Visible staining to gain qualitative confirmation of unknown materials |
0.3 µm to 1 mm |
The accuracy of equipment used to determine particle size distribution is verified by calibration with reference particles of known concentration and size traceable to the International System of Units.
The concept of three-dimensional particle size is critical to understanding how particle standards interact with the various types of measurement equipment and how to compare the results across methodologies. Different techniques use different measurements and algorithms to determine size (e.g., volume equivalent diameter, surface equivalent diameter, and drag or Stokes diameter).
SIZE AND COUNT DISTRIBUTION
Light Obscuration (working range 1–300 µm)
principle of operation
The sample is passed between a light source and a sensor. Counts are generated when individual particles pass between the two, disrupting the light, which yields a voltage spike. The height of each voltage spike depends on the size of the particle causing it. The particle size to be recorded is generated from a size–voltage response calibration curve constructed using monosphere, certified reference size standards (typically polystyrene beads). LO tabulates particle size as the diameter of a circle having an equivalent cross-section. The product may be sampled directly from the container, pooled from several containers, or prepared as a dilution (see general chapters Globule Size Distribution in Lipid Injectable Emulsions 729, 788, Particulate Matter in Ophthalmic Solutions 789, and Methods for the Determination of Particulate Matter in Injections and Ophthalmic Solutions 1788).
Advantages
-
Primary USP–NF method with large archive of historical data
-
Particles directly observed in solution
-
Measurements are straightforward and quick
-
Capable of unit–unit sampling; analyzes almost entire sample
-
Easily implemented
-
Medium-to-high sample throughput.
Limitations
-
One sensor head cannot cover the full working range
-
Tabulates particle size as the diameter of a circle having an equivalent cross-section
-
Dilution of the sample may be required and may change sample properties
-
Upper limits for particle load that can be measured depend on sensor used and potential for coincidence counting
-
May underestimate subvisible protein particles formed in formulation because of low contrast
-
Medium should be transparent for laser light
-
Sensitive to air bubbles, and some degassing techniques can change sample properties
-
Destructive, i.e., sample cannot be reused.
Electrical Sensing Zone (Coulter Principle) (working range 0.4–1600 µm)
principle of operation
The sample is diluted into an electrolyte solution and drawn through a small aperture, passing between active electrodes, interrupting an electric field. Response is based on the displacement volume of the electrolyte, and thus size is reported as the equivalent spherical diameter (the diameter of a calibration standard sphere of the same volume as the particle). The response is unaffected by particle type (e.g., variations in color, hardness, opacity, refractive index).
Advantages
-
Determines particle size in terms of equivalent spherical volume
-
Not affected by optical properties of the solution
-
Orthogonal technique to light-based methods, i.e., LO
-
Medium-to-high sample throughput for a single orifice.
Limitations
-
Low particle concentration required
-
Each orifice has limited measuring range (2%–60% of orifice diameter)
-
Near the lower end of the working range, electrical noise may be hard to distinguish from true particle counts
-
For broad particle size distribution, more than one orifice may be necessary, which will involve reduced sample throughput
-
Sample must be diluted in electrolyte solution if the formulation buffer does not have sufficient conductivity; the dilution may alter sample properties
-
Smaller orifices require higher conductivity of medium and can require significantly increased buffer concentration
-
Destructive, i.e., sample cannot be reused.
Laser Diffraction (working range 0.1–3500 µm)
principle of operation
Laser diffraction detects particles passing through a laser beam as they scatter light at an angular distribution of intensity that depends on the size of the particles. Therefore, it is possible to calculate particle size distributions if the intensity of light scattered from a sample is measured as a function of angle. This angular information needs to be compared with a scattering model (Mie theory) to calculate the size distribution.
Advantages
-
Brief analysis time (up to 30 s)
-
Analytical samples can be retrieved
-
Limited sample preparation
-
Scattering pattern allows molecular mass calculation
-
High sample throughput
-
Large dynamic range.
Limitations
-
Intensity is directly proportional to size, which can lead to misinterpretation of the population
-
Analysis relies on a theoretical fit that requires refractive index and other optical factors
-
Medium should be transparent for laser light
-
Assumes particles are spherical and not interacting with each other
-
Low concentration of particles required; dilution may be necessary and may change sample properties
-
Complications can occur when comparing data from one instrument vendor to another
-
Particle–particle interactions and occasionally multiple scattering may yield apparent size results that differ significantly from actual particle size
-
Poor results for polydispersions when small and large particles are presented simultaneously
-
For exact molecular weight calculation, monodisperse particles are required.
SIZE AND MORPHOLOGY
Light Microscopy (LM) (working range 0.3 µm to 1 mm)
principle of operation
Optical or light microscopy involves passing light through a series of lenses after it is transmitted through or reflected from the prepared sample. The light is detected directly by the eye, imaged on a photographic plate, or captured digitally. Samples can be analyzed directly or after filter capture. Light microscopy can be combined with static imaging analysis, thereby allowing digital images to be captured and further deconvolved using the software systems in image analysis. When using image analysis, one may select certain parameters to screen out unwanted artifacts or particle populations (circularity, aspect ratio, and others) and then begin studying the selected population set (see Optical Microscopy 776, 788, 789, and 1788).
Advantages
-
Direct observation of particles in product fluid and filter capture
-
Visualization, counting, sizing, and morphological characterization of particles are readily obtained
-
Direct observation of particle may provide immediate recognition and identification
-
Can be coupled with spectroscopic analyses (infrared, Raman) to identify chemical composition
-
Particle identification can be enhanced via image database search and comparison
-
Only small sample volume is needed for direct observation
-
Membrane filtration is secondary to the 788 LO method.
Limitations
-
Time consuming
-
Low sample throughput
-
Limited depth of field at high magnification can affect sizing accuracy
-
Only a small sample volume is analyzed, so sampling must be done carefully
-
For filter-captured samples, protein particles can pass through membranes and may present as amorphous background
-
For filter-captured samples, potential exists for sample preparation artifacts
-
Difficult to visualize particles with low optical contrast; must use phase, differential interference contrast microscopy, polarized light microscopy, and other methods
-
Destructive, i.e., sample cannot be reused for filter-captured samples.
Flow Imaging Analysis [working range 0.7–100 µm (size) and 4–100 µm (morphology)]
principle of operation
This method captures digital images of materials within a flowing sample stream and uses post-collection analyses of the stored images. As in optical microscopy, which is essentially a static image analysis system, dynamic image analysis or flow microscopy captures multiple separate images, in this case using a dynamic, flowing system. Dynamic image analysis uses the components of illumination, objective lens, and focusing lens of a microscope, and adds a closed fluid pathway within a defined dimensional flow cell, plus camera and processor for acquisition and analysis. Thus, the method provides in situ conditions with realistic views of the particles. Visualization of the particles of gas, liquid, semisolids, or solids with this technique aids the interpretation of other in-suspension measurements that cannot visualize the particles being studied. The particles are moving and are likely tumbling, and this must be accounted for in the system design or data consideration. Increasing the system magnification and pixel resolution and reducing the flow cell depth will augment the quality of the captured image. An additional advantage of dynamic flow microscopy is the ability to apply size and feature analyses to the particle image. Also, any number of additional algorithms may be applied to study the collected image set as necessary. Particle image clarity, contrast, and shape may indicate possible source or type of particle, and with sufficient comparator data, mathematical filters can be applied to analyze and quantify different populations in a sample. This approach has been used to help distinguish silicone oil from protein aggregates.
Advantages
-
Direct observation of particles in product fluid (in situ)
-
Analysis of captured digital images allows visualization, counting, sizing, and characterization of particles
-
Retained images may be further analyzed by different algorithms
-
Actual particle images facilitate understanding of contamination sources and product nature
-
Compared to LO or membrane microscopy (MM), increased sample volume and reduced analysis time
-
Morphology and relative refractive index of particles apparent, easily calibrated
-
Technique can be used with image databases to identify some particles
-
High repeatability, high resolution, and good selectivity
-
Medium-to-high sample throughput.
Limitations
-
Minimum particle size necessary
-
Results dependent on algorithm used to select, classify, and size particles
-
Difficult to visualize and appropriately quantify number and size of particles with low optical contrast, such as translucent proteins
-
Dilution of sample may be required and may change sample properties
-
Destructive, i.e., sample cannot be reused.
Electron Microscopy (working range Angstroms to mm)
principle of operation
In EM, a beam of electrons is used to illuminate the samples and produce a magnified image. The practical spatial resolution limit of EM for biological samples is approximately 1 nm, which greatly exceeds resolution requirements for typical >0.1-µm particles in protein solutions. For EM analysis, subvisible particles should be either individually isolated or captured on a suitable filter and then mounted onto special grids or stabs for observation in the microscope. Traditional EM operates in high vacuum (about 10–6 Torr). Sample drying and coating is a required preparation step. Newer technologies allow examination of uncoated, as-is samples (environmental or wet EM). Detection techniques are divided as follows: (a) scanning electron microscopy (SEM), which may use secondary electron or back-scattering signals; (b) transmission electron microscopy (TEM), which visualizes electrons penetrated through sample; and (c) scanning tunneling electron microscopy (STEM), which combine SEM and TEM principles (see also Scanning Electron Microscopy 1181).
Advantages
-
Visualization, counting, sizing, and morphological characterization of particles are readily obtained
-
Direct observation of particle may provide immediate recognition and identification
-
High repeatability, high resolution, and good selectivity
-
Information about chemical composition of object can be obtained using energy-dispersive spectroscopy (EDS) or electron energy loss spectroscopy (EELS) (see below)
-
Enhanced depth of field compared with photon methods
-
Image analysis and building permits quasi-3D imaging of particles.
Limitations
-
Time-consuming preparation and analysis
-
Low sample throughput
-
Possible changes in structure and size may arise from sample preparation and high-vacuum operation
-
Specialized sample preparation requirements may exist, depending on the nature of the sample
-
Contrast staining with heavy metals salts may be required for enhancing protein samples
-
Protein particles are flexible and can pass through membranes
-
Destructive, i.e., sample cannot be reused.
CHARACTERIZATION
Characterization of the protein active ingredient includes not only size distribution assessment but also determination of the chemical nature of the primary protein as well as conformation, aggregation, and self-associated species (see Table 1). The size of the fundamental particle–aggregate is important; however, the nature of the solids and the association with other species are similarly important.
Fourier-Transform Infrared (FTIR) Microspectroscopy (working range 10 µm to 1 mm or greater)
principle of operation
When light interacts with a substance, the light can be reflected, absorbed, or scattered. Infrared spectroscopy determines the vibrational properties of matter, typically in the 200–4000 cm–1 range, by analyzing the interaction with light while detecting the absorption of photons. In addition, the use of microscopy-based instruments, thus microspectroscopy, allows selection of individual particles or domains of interest, with the benefit of filters and polarizers for microscopes and the ability to capture an image, along with vibrational or other imaging data. Vibrational spectroscopy techniques are particularly useful for analysis of particles in protein solutions.
Particles can be isolated individually or captured on a membrane filter and then analyzed using FTIR microscopy. Because the infrared signal from water interferes strongly with protein bands, these samples usually require thorough drying before analysis. Typically, FTIR microscopes can operate in either image mode or spot mode. In image mode, the infrared signals are collected from individual pixels in the pre-defined area. However, spot mode usually provides higher signal-to-noise ratio, because the combined infrared signal is collected from the area defined by the aperture size. Many organic compounds have unique infrared signatures. Spectra collected for an individual particle can be analyzed by performing an infrared spectra library search or by comparing the particle’s spectra results with those of suspected materials. See Spectrophotometry and Light-Scattering 851 for more information.
Advantages
-
Microspectroscopy has the same sample advantages as LM
-
FT instruments excel compared with dispersive spectrometers (grating monochromator) for particle analysis
-
Provides rapid chemical identification and classification of particles, manually or automatically, in comparison with spectral databases
-
Identification, structure, and composition can be derived from spectral data
-
Provides some information on protein structure
-
Nondestructive; samples can be reused.
Limitations
-
May have the same limitations as LM
-
FTIR provides strong absorption for common materials, such as silicones, water, and glass, which may interfere with or hinder sample analysis
-
Identification depends on availability of information on possible components in spectral libraries
-
Particle size limit is dependent on instrument resolution (~10 µm for FTIR)
-
Process of particle identification becomes time consuming, if not available in a database
-
FTIR is not sensitive to many inorganic materials or other molecules that do not possess a dipole moment
-
Certain detectors are not sensitive to inorganic materials.
Dispersive-Raman Microspectroscopy (working range 0.5 µm and greater)
principle of operation
As with infrared spectroscopy, Raman spectroscopy is the study of how light interacts with a substance. A defined, monochromatic photon source (laser) is focused on the sample, thereby producing reflectance, absorption, and scatter, probing the same vibrational states probed by infrared microspectroscopy. Specifically, Raman spectroscopy is the study of the inelastically scattered photons (Raman scattering). Photons are also scattered elastically (Rayleigh scattered) with no change in wavelength, but are of use only to mark the laser excitation energy. A virtual excitation state is attained, which then relaxes to a base vibrational or rotational state, emitting energy characteristic of a functional group such as molecular and related atoms. Lower (Stokes) and higher (anti-Stokes) shifts are recorded due to interaction with the electron cloud of the functional group bonds. Lasers are used because only a very small proportion of Raman-scattered photons (only about 1 in 106–108) exhibit wavelength shift. Raman microspectroscopy combines a light microscope with a coincident laser and white light pathway. The microscope, with the benefit of filters and polarizers, aids in selection of individual particles or domains of interest to allow capturing of an image, along with vibrational spectra.
Fourier-Transform Raman is generally complementary to dispersive Raman, and in using longer wavelength excitation yields little sample fluorescence. One important advantage of Raman microscopy compared with infrared is the ability to analyze aqueous samples directly, often in glass sample holders. Individual particles can be analyzed in situ, and in some cases directly in the container, if it does not interfere with the Raman signal. Similar to the infrared analysis, unknown spectra can be analyzed either by a spectral library search or by matching the spectra of individual reference compounds.
Raman spectroscopy theory and techniques are further described in Raman Spectroscopy 1120.
Advantages
-
Same sample advantages as LM
-
Dispersive instruments excel over Fourier-Transform spectrometers for particle analysis
-
Rapid chemical identification and classification of particles, manually or automatically by comparison with spectral databases
-
Identification, structure, and composition can be derived from spectral data
-
Provides some information on protein structure
-
Good lower size limit; instrument resolution is ~0.5 µm for dispersive-Raman technique
-
Raman analysis viable for many organic and most inorganic compounds
-
Identification uses Raman shift data, known for molecular categories and functional groups
-
Nondestructive; samples can be reused
-
Glass and plastic packaging have weak Raman spectra, thus interference from packaging is minimized.
Limitations
-
Same limitations as with LM
-
Specimen fluorescence may hinder data collection
-
With sensitive samples, there can be laser-induced changes (heat, light)
-
Many organic species yield no Raman shift; that is, there is no molecular polarization
-
Identification may depend on the information available in proprietary and public spectral libraries.
Electron Microscopy (EM) with Energy-Dispersive X-Ray Spectrometry (EDS or EDX) (working range for qualitative analysis, 0.1- to 3-µm particles; for semiquantitative analysis, >3-µm particles)
principle of operation
Energy-dispersive X-ray spectrometry (EDS or EDX) is based on characteristic X-ray photon emission generated from a specimen through interaction with a high-energy source, such as an electron beam. Typically, an elemental intensity is plotted against X-ray emission energy, which correlates with the content of atomic valence shell transition energies. Current instrument technology uses thin-window and windowless detectors positioned near the sample in EM systems, which improves detection and signal-to-noise ratio for all elements, especially lighter elements, e.g., Z < 11 (sodium). Immediate, qualitative elemental composition is attained for solid specimens (see also 1181 for general EM and related EDS background information).
Advantages
-
Complementary to EM and available for most commercial electron microscopes
-
Determines elemental composition of the sample
— Rapid qualitative collection for small particles— Semiquantitative analysis (1% LDL and ±20% RSD) possible in controlled experiments
-
Most useful during survey investigation of particle origin
-
Involves manual elemental screening and/or search of databases
-
Elemental mapping can assist in protein particle visualization.
Limitations
-
Not all instruments are equipped for determination of light elements (e.g., Z < 11)
-
Quantitative analysis entails measuring line intensities for each element in the sample and for the same elements in calibration standards of known composition
-
Strong background may be contributed by filter membrane material and sample coating.
Electron Microscopy (EM) with Electron Energy Loss Spectroscopy (EELS) (working range 0.5 µm to 1 mm)
operation principle
Electron energy loss spectroscopy (EELS) measures the vibrational motion of atoms and molecules on and near the surface by analyzing the energy spectrum of low-energy electrons back-scattered from it.
Advantages
-
Used in conjunction with transmission electron microscopy
-
Determines elemental composition of the sample, especially light elements (e.g., Z < 11)
-
Can provide information about chemical bonding
-
Qualitative and quantitative analyses are possible
-
Has lower detection limits than in EDS.
Limitations
-
Difficult to interpret
-
Strong background
-
Quantitative analysis entails measuring line intensities for each element in the sample and for the same elements in calibration standards of known composition.
Time-of-Flight Secondary Ion Mass Spectrometer (TOF-SIMS) (working range µm to near mm)
operation principle
Time-of-Flight Secondary Ion Mass Spectrometer (TOF-SIMS) is a surface-sensitive analytical method that uses a pulsed ion beam [microfocused cesium (Cs) or gallium (Ga)] to remove molecules from the very outermost surface of the sample. The particles are removed from atomic monolayers on the surface (secondary ions). These particles are then accelerated into a “flight tube”, and their mass is determined by measuring the exact time at which they reach the detector (i.e., time-of-flight).
Advantages
-
Surveys of all masses on material surfaces; these may include single ions (positive or negative), individual isotopes, and molecular compounds
-
Elemental and chemical mapping on a submicron scale
-
High mass resolution, to distinguish species of similar nominal mass
-
High sensitivity for trace elements or compounds, on the order of ppm to ppb for most species
-
Surface analysis of insulating and conducting samples
-
Depth profiling (in the near surface environment, on the order of individual atomic layers to 10 s of nanometers)
-
Nondestructive analysis
-
Retrospective analysis, for post-data acquisition analysis and interpretation of stored images and spectra
Limitations
-
Generally does not produce quantitative analyses (semi-quantitative at best)
-
Optical capabilities typically limited, making it difficult to find grains or specific regions of interest for analysis
-
Charging may be a problem in some samples, although charge compensation routines are generally sufficient to overcome these problems
-
There is commonly an image shift when changing from positive to negative ion data collection mode, making it difficult to collect positive and negative ion data on exactly the same spot.
Staining Assay (working range 0.3 µm to 1 mm)
operation principle
Staining methods utilize reagents to provide specific reactions, such as visible staining or fluorescence, which provide qualitative confirmation of an unknown materials category or identity. Generally conducted via microscopical methods; microscopy advantages and limitations apply.
Advantages
-
Direct observation of staining effects upon particles in product fluid and by filter capture
-
Visualization, counting, sizing, and morphological characterization of particles are readily obtained
-
Direct observation of particle may provide immediate recognition and identification
-
Can be coupled with spectroscopic analyses (infrared, Raman) to identify chemical composition
-
Surface analysis of insulating and conducting samples
-
Particle identification can be enhanced via image database search and comparison
-
Only small sample volume is needed for direct observation
Limitations
-
Low sample throughput
-
Limited depth of field at high magnification
-
Only a small sample volume is analyzed, so sampling must be done carefully
-
For filter-captured samples, protein particles can pass through membranes and may present as amorphous background
-
For filter-captured samples, potential exists for sample preparation artifacts
-
Difficult to visualize particles with low optical contrast; must use phase, differential interference contract microscopy, polarized light microscopy, and other methods
-
Destructive; that is, sample cannot be reused for filter-captured samples
STRATEGY
Particles in therapeutic protein products generally have continuous size distributions with exponentially higher numbers of the smaller (<10 µm) particles. Information generated from analysis of inherent protein particles can be used for guiding development efforts and to facilitate communication and understanding of this important quality attribute of therapeutic protein products. A rational strategy should be developed for characterizing protein aggregates or particles. The categories in Table 1 provide a guideline for differences observed between types of protein aggregates, recognizing that size distribution analysis of these species is important, but it is not the only informative characteristic. Describing the characteristics of the protein aggregates in the manner shown in Table 1 can help define the population and/or characteristics that may be the most important for assessment of the effect on product quality.
To obtain a comprehensive characterization of the particle distribution, a variety of analytical techniques must be applied. There are a number of techniques available to measure and characterize particles, each with its advantages and limitations. Multiple methods are needed because they differ in their ability to analyze specific attributes of the particle. By using orthogonal methods, one can better ascertain the nature of the particles (extrinsic, intrinsic, or inherent), the size distribution, and other aspects that may allow differentiation between particles. Information about the particle characteristics can be used to perform a risk assessment around the presence of aggregates and to help identify potential mechanisms of formation. This knowledge about the particles' characteristics can also be used during formulation and process development to inform choices of the formulation and process steps, which can help in reducing the overall particle content of the drug product.
A comprehensive strategy for assessing subvisible particles involves multiple phases from development through commercialization and during marketing. Particle measurement/characterization may begin as early as candidate selection, with the use of high-throughput techniques to predict particulate-forming tendencies of the molecules under consideration. This involves using small amounts of material and providing a relative ranking of aggregation propensity.
Early Development
At this stage, the focus is on understanding the typical particle profile for the product, including the size distribution and the characteristics of the aggregates most commonly seen in the drug product. The particle size and count in the 1- to 100-µm range should be monitored at the time of manufacture and during stability.
Late Development
At this stage, the focus is on understanding the drug product and comparability between lots, as well as the linkage between particle size analysis and formulation, manufacturing, and use. Particle size and count in the 1- to 100-µm range should be monitored in clinical batches. Enhanced characterization of aggregates and particles, including pivotal product batches, should be performed. Quantitative and qualitative data about the subvisible particles formed under storage, use, and stress conditions should be collected, and risk mitigation strategies created. When the final method for monitoring the product’s particle size and count is chosen, control strategies for inherent and intrinsic particles should be established. The overall control strategy developed may include action limits beyond which an investigation may be warranted.
Marketing
At this stage, one should collect data over commercial batches to align with the overall control strategy, which may include the sub-10-µm (1- to 10-µm) range. Use quantitative as well as qualitative analysis during the management changes for the post-marketing life cycle. When an investigation occurs, the knowledge obtained during development can be used to design mitigation strategies. Stability studies are also part of life cycle management, and are addressed later in the Application section.
Overall Perspective
In-depth characterization of aggregates during development, coupled with correlation of results from these tests to those used for product release (such as LO), may provide the basis for a rational, overall control strategy. Aggregates are usually present in a continuum of size from oligomers to particles that are hundreds of micrometers. If a correlation (or mathematical relationship) can be demonstrated in the counts of particles from oligomers through the >25-µm sizes assessed as part of the compendial method, it might be possible to rely on the LO determination of the >10- and >25-µm particle content to reveal the effect of changes and trends in manufacturing on 1- to 10-µm particles. In this situation, the overall control strategy would be used to establish action limits for particles >10 µm, which when exceeded would trigger an investigation, including the characterization methods discussed in this chapter.
SAMPLE CONSIDERATIONS
Certain aspects of sample handling and volume require special consideration. Handling and degassing procedures need to be developed and applied to avoid artifacts such as false positives attributable to bubbles, or aggregates generated during the sample preparation steps. Under certain circumstances, dilution of samples may be necessary to obtain reliable results. Aspects of sample handling are described in 787.
APPLICATION
A key focus during development of a protein drug product is to gain knowledge about the nature, source, and number of inherent particles/aggregates present in the therapeutic formula, as well as their stability and overall effects on product quality, safety, and efficacy.
Protein aggregates in the 1- to 10-µm size range may also carry a risk of potentiating immunogenicity against the therapeutic activity (4,5). From a product-development perspective, changes in the concentrations of proteinaceous subvisible particles, particularly in the 1- to 10-µm range, can be an early and sensitive indicator of potential issues with product stability (6,7). Apart from stability, particles in these size ranges should also be measured as part of the risk assessment exercise to improve understanding of how various stressors and conditions of use affect the product. These conditions could include thermal, freeze/thaw, light, transport, dilution/admixture, and use characteristics. Particle counts in the 1- to 10-µm range tend to be significantly (orders of magnitude) greater than those in the  10-µm range. This makes the 1- to 10-µm range useful for the purpose described above, because changes can be easier to observe. However, the counts also tend to be quite variable, and thus the ability to discern trends requires proper controls, adequate sampling, and good technique.
10-µm range. This makes the 1- to 10-µm range useful for the purpose described above, because changes can be easier to observe. However, the counts also tend to be quite variable, and thus the ability to discern trends requires proper controls, adequate sampling, and good technique.
Detailed characterization of subvisible particles, especially inherent particles, provides information that can support the formulation and product development program, leading to a robust process and commercial control strategy. On the other hand, although characterization alone may not reveal possible correlations between an immune response and aggregates/particles, the use of multiple methods applied to multiple drug product lots helps inform the definition of product quality, which can be linked to safety and efficacy data obtained from clinical trials. This knowledge is also useful in life cycle management, such as movement of product between manufacturing sites, changes in presentation (liquid to lyophilized; vial to prefilled syringe), and changes in strength.
To illustrate these points, two examples are presented below; they show ways that complementary techniques can be used to gain product knowledge.
Example 1: Comparison of Drug Product Configurations
Not all drug products are formulated as liquids. In one case of a lyophilized drug product filled in glass vials, a liquid formulation in a prefilled syringe configuration was developed to facilitate patient administration. A comparison of subvisible particle counts by LO was expected to show that the particle count of the reconstituted lyophilized formulation was higher than in the liquid formulation, but the opposite results were observed, with the liquid formulation containing significantly higher levels of particles >2 µm [~130,000 particles per container (ppc) compared to ~30,000 ppc]. These samples were then assessed by the MM method, and the results were more consistent with the expected outcome: for particle sizes >5 µm, the lyophilized formulation contained ~70 ppc compared to ~14 ppc for the liquid formulation. Analysis was then performed using flow microscopy to understand the discrepancy in results between the MM and LO methods. Flow microscopy supported the LO results, with the liquid formulation showing higher numbers of particles >2 µm (~600,000 ppc) compared to the lyophilized formulation (~150,000 ppc). Examination of the structure of the particles revealed a different morphology in the liquid formulation, with a predominant number of the particles exhibiting a spherical shape, consistent with the presence of silicone oil droplets from the prefilled syringe container. These results were confirmed by testing a sample of the liquid formulation that had not been exposed to the prefilled syringe (the source of the silicone oil), which showed <20,000 ppc. Once the silicone oil droplets were accounted for, the number of remaining particles in the liquid formulation in the prefilled syringe was lower than in the lyophilized formulation, consistent with the MM results.
Example 2: Selection of Filling Technology
Filling processes can affect product quality by introducing physical stresses such as adsorption, shear, friction, and cavitation, which may lead to protein aggregation. Certain drug product filling pumps may shed extrinsic particles that can lead to heterogeneous nucleation-induced aggregation.
The effect of using a stainless steel displacement piston pump on the subvisible particulate content of solutions was investigated using multiple techniques to analyze the particulates in solution (8). Coulter counter analysis of a buffer solution passed through the pump revealed that the pump shed particles in solution, with the pumped solution containing >13,000 particles per mL in the 1.5- to 6-µm range. Elemental analysis confirmed that the particles were stainless steel. Analysis of the particle size distribution in this solution, performed using a laser diffraction particle sizer, showed that most of the particles were between 0.25 and 0.95 µm. Incubation of stainless steel nanoparticles with a solution of MAb-Y led to increases in both the number of particles and the particle size distribution over time. Characterization of the particles by FTIR spectroscopy showed that the protein adsorbed onto the metal particles contained a slightly perturbed secondary structure compared to the protein in solution. It was concluded that the metal particles served as nucleation sites for protein aggregation. In this case, selection of a different type of filler may be warranted to mitigate protein aggregation.
A comparison of the effect of different pump types on subvisible particulate formation in a solution of MAb-Z was performed (9). Flow microscopy showed that piston pumps, made of either stainless steel or ceramic, produced particles in the size range of 1–100 µm at levels approximately 100-fold higher than the control samples. Examination of the microscopic images of these particles revealed that the particles were translucent, suggesting they were protein based. Incubation of the protein solution with shed particles from the piston pumps did not lead to a significant increase in either the particle size distribution or particle counts for these solutions, indicating that extrinsic particles did not serve as nucleation sites for the protein. In this case, the formation of particles in the solution was considered to be from shear stress caused during the pumping operation. These results were supported by examination of other filler types (peristaltic, time-pressure, and rolling diaphragm), which produced solutions with significantly lower particle load.
SUMMARY
This chapter provides strategies for describing, characterizing, and identifying the particle content in therapeutic protein injections, including extrinsic and intrinsic particle population. Information regarding specific methods that can be used for these purposes, as well as their advantages and limitations, is discussed. A rational, comprehensive control strategy can be developed on the basis of an in-depth characterization of aggregates during development, coupled with correlation of results from these tests to those used for release. Aggregates are usually present in a continuum of sizes, from oligomers to particles that are hundreds of micrometers. Correlation in the counts of particles from oligomers through the >25-µm size, assessed as part of the compendial method, may make it possible to rely on the LO determination alone to reveal changes and trends in manufacturing. In this situation, the overall control strategy would be used to establish action limits for particles >10 µm, which when exceeded would trigger an investigation, including the characterization methods discussed in this chapter.
REFERENCES
-
Narhi LO, Schmit J, Bechtold-Peters K, Sharma D. Classification of protein aggregates. J Pharm Sci. 2012; 101(2):493–498.
-
Felsovalyi F, Janvier S, Jouffray S, Soukiassian H, Mangiagalli P. Silicone-oil-based subvisible particles: their detection, interactions, and regulation in prefilled container closure systems for biopharmaceuticals. J Pharm Sci. 2012; 101(12): 4569–4583.
-
Ripple DC, Wayment JR, Carrier MJ. Standards for the optical detection of protein particles. Am Pharm Rev. 2011; July Issue. http://www.americanpharmaceuticalreview.com/Featured-Articles/36988-Standards-for-the-Optical-Detection-of-Protein-Particles/. Accessed 10 January 2013.
-
Carpenter JF, Randolph TW, Jiskoot W, Crommelin DJ, Middaugh CR, Winter G, et al. Overlooking subvisible particles in therapeutic protein products: gaps that may compromise product quality. J Pharm Sci. 2009; 98(4):1201–1205.
-
Singh SK, Afonina N, Awwad M, Bechtold-Peters K, Blue JT, Chou D, et al. An industry perspective on the monitoring of subvisible particles as a quality attribute for protein therapeutics. J Pharm Sci. 2010; 99(8):3302–3321.
-
Barnard JG, Singh S, Randolph TW, Carpenter JF. Subvisible particle counting provides a sensitive method of detecting and quantifying aggregation of monoclonal antibody caused by freeze-thawing: insights into the roles of particles in the protein aggregation pathway. J Pharm Sci. 2011; 100(2):492–503.
-
Cao S, Jiang Y, Narhi L. A light obscuration method specific for quantifying subvisible particles in protein therapeutics. Pharmacopeial Forum. 2010; 36(3):824–834.
-
Tyagi AK, Randolph TW, Dong A, Maloney KM, Hitscherich C, Jr, Carpenter JF. IgG particle formation during filling pump operation: a case study of heterogeneous nucleation on stainless steel nanoparticles. J Pharm Sci. 2009; 98(1):94–104.
-
Nayak A, Colandene J, Bradford V, Perkins M. Characterization of subvisible particle formation during the filling pump operation of a monoclonal antibody formation solution. J Pharm Sci. 2011; 100(10):4198–4204.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Desmond G. Hunt, Ph.D.
Senior Scientific Liaison (301) 816-8341 |
(GCDF2010) General Chapters - Dosage Forms |
USP38–NF33 Page 1680
Pharmacopeial Forum: Volume No. 39(6)