



Change to read:
Change to read:
-
Fluid pathways of catheters and administration sets such as solution administration sets, extension sets, transfer sets, blood administration sets, intravenous catheters, implants, extracorporeal oxygenator tubings, dialysis tubing, intramuscular drug delivery catheters, and transfusion and infusion assemblies
-
Liquid medical devices such as dialysate
-
Implantable medical devices such as heart valves and vascular grafts, other medical devices with a nonpyrogenic claim that may come into contact with blood or cerebrospinal fluid
-
Gels with a nonpyrogenic claim including demineralized bone matrices and drug delivery systems
Endotoxin limit
K = limit for each device
N = number of devices tested
V = total volume of the extract
-
The endotoxin limit for the finished device is NMT 20 USP Endotoxin Units per device and NMT 2.15 USP Endotoxin Units for devices in contact with cerebral spinal fluid.
-
For devices that directly or indirectly contact the intraocular environment, a lower endotoxin limit may apply.
-
The endotoxin limit for the device extract in Endotoxin Units/mL is calculated as:
(K × N)/V
K = limit for each device
N = number of devices tested
V = total volume of the extract
Kit:
Table 1. Selection of Product Units for Testing
| Product Unit
|
Item for Testing |
|---|---|
| Individual medical device in primary package where each medical device is used individually in clinical practice |
Individual medical device |
| Set of components in a primary package where components are assembled as a product and are used together in clinical practice |
Combination of components |
| Number of identical medical devices in one primary package where each medical device is used independently in clinical practice |
Single medical device taken from the primary package |
| Kit of procedure-related medical devices where each medical device is used independently in clinical practice and where each medical device may have a different endotoxin limit |
Each type of medical device that has a nonpyrogenic claim, or All items together |
Lambda (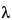 ):
):
Product families:
Extracting or rinsing solution:
 85
85 , Water for Bacterial Endotoxins Test (BET)] or other suitable solvent demonstrated to be noninterfering with the BET assay is used to extract or rinse the medical device.
, Water for Bacterial Endotoxins Test (BET)] or other suitable solvent demonstrated to be noninterfering with the BET assay is used to extract or rinse the medical device.
Sampling
-
Sample—A sample for end-product testing must be in its final configuration and packaging, including all component parts that make up the final medical device.
-
Representative sampling—A representative sample is a sample plan that is created and justified based on the assumption that the manufacturing process is validated and in a state of control.
-
Sample size—The number of devices chosen for routine testing is dependent on the size of the lot, level of control, statistical considerations, and historical performance. In most cases, each lot of product must be tested using an appropriate number of samples, NMT 10, taken at random to represent the quality of the lot. Alternate sampling plans that utilize small sample sizes or do not test each lot of product must be clearly defined and must be supported/justified by a robust risk assessment.
-
Pre-post sterilization samples—When qualifying a BET assay for pre-sterilization samples, equivalency in test results must be shown with post-sterilization samples to ensure that sterilization and post-sterilization handling have no adverse impact on the accuracy of the test result. Pre-sterilization testing is inappropriate for products that support microbial growth.
Suitability testing (also known as test for interfering factors):
Apparatus:
Depyrogenate all glassware and other heat-stable materials in a hot air oven using a validated process. If employing plastic apparatus such as microplates and pipet tips for automatic pipetters, use apparatus that is shown to be free of detectable endotoxin and does not interfere with the test.
Analysts must be properly trained to perform the assay.
The assay sensitivity must be confirmed as described in 85 sections Test for Confirmation of Labeled Lysate Sensitivity for gel clot reagents and Assurance of Criteria for the Standard Curve for quantitative methods. Assay sensitivity confirmation must be carried out when a new batch of reagent (lysate or endotoxin) is used or when there is any change in the test conditions that could affect the outcome of the test.
Endotoxin limit:
K = amount of endotoxin allowed per device (20 Endotoxin Units per device; 2.15 for intrathecal devices)
N = number of devices tested
V = total volume of the extract or rinse
(K × N)/V
K = amount of endotoxin allowed per device (20 Endotoxin Units per device; 2.15 for intrathecal devices)
N = number of devices tested
V = total volume of the extract or rinse
Note—The volume of the rinse or extract may be adjusted for the size and configuration of the device.
The standard extracting fluid for extracting, rinsing, or soaking medical devices is Water for BET (see 85). If necessary, other solvents may be used after it has been demonstrated that they do not interfere with the performance of the assay. Either the entire device if claimed to be nonpyrogenic or all components of the device that are claimed to be nonpyrogenic, including fluid pathways, must come in contact with the extraction fluid for the entire course of the extraction.
The standard extraction method is to soak or immerse the device or flush the fluid pathway with extracting fluid that has been heated to 37 ± 1.0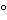 , keeping the extracting fluid in contact with the relevant surface(s) for NLT 1 h. Alternate extraction or rinsing methods may be used, but must be demonstrated to be equivalent or better than the standard method. Extracts from individual medical devices are typically pooled for testing.
, keeping the extracting fluid in contact with the relevant surface(s) for NLT 1 h. Alternate extraction or rinsing methods may be used, but must be demonstrated to be equivalent or better than the standard method. Extracts from individual medical devices are typically pooled for testing.
Suitability:
= sensitivity of the test method in Endotoxin Units/mL
For the suitability test, medical device extracts are considered to be the sample solutions under test and are tested as outlined in 85 for the chosen technique in the section Test for Interfering Factors.
If necessary, adjust the pH of the solution to be examined (or dilution thereof) so that the pH of the mixture of the lysate and sample solution falls within the pH range specified by the lysate manufacturer, usually pH 6.0–8.0. The pH may be adjusted by use of an acid, base, or suitable buffer as recommended by the lysate manufacturer. Acids and bases may be prepared from concentrates or solids with Water for BET (see 85) in containers free of detectable endotoxin. Buffers must be shown to be free of detectable endotoxin and interfering factors.
If the undiluted rinsing or extracting solution is determined to be unsuitable for 85, the test for interfering factors may be repeated utilizing dilution, or a method that will neutralize or eliminate the interfering substance.
If glucan interference is suspected, the use of a glucan blocking agent or endotoxin-specific lysate is permissible.
Dilution in Water for BET (see 85) or other validated solvent by a factor not to exceed the maximum valid dilution (MVD) is permissible. The MVD for medical devices is the following:
MVD = Endotoxin limit (of the rinsing or extracting solution in Endotoxin Units/mL)/
Interference may be overcome by suitable treatment of the device extract. Methods for overcoming interference and the qualification of such treatments are described in 85. To establish that the chosen treatment effectively eliminates interference without loss of endotoxins, perform the assay described in 85 using the preparation to be examined to which standard endotoxin has been added and which has then been submitted to the chosen treatment.
Changes to extraction techniques require a new suitability study. Because of possible changes in interference patterns between standard BET methods, a change in BET methodology (e.g., from gel clot limits test to kinetic chromogenic) requires a re-execution of the suitability study.
Routine testing:
Routine testing is to be performed as outlined in 85, following instructions for incubation and controls listed under the chosen assay technique.
Interpretation of test results:
If a device fails to meet this requirement, an investigation may be initiated per documented procedures. A failed, valid BET assay may not be repeated using Pyrogen Test 151 in its place.
-
ANSI/AAMI ST72: 2011, Bacterial endotoxins—Test methods, routine monitoring, and alternatives to batch testing. Association for the Advancement of Medical Instrumentation, Arlington, VA.
-
United States Food and Drug Administration, Guidance for industry pyrogen and endotoxins testing: questions and answers, June 2012. http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm314718.htm
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Radhakrishna S Tirumalai, Ph.D.
Principal Scientific Liaison (301) 816-8339 |
(GCM2010) General Chapters - Microbiology |
USP38–NF33 Page 212
USP38–NF33 Supplement : No. 1 Page 7039
Pharmacopeial Forum: Volume No. 40(3)