



This informational chapter provides a general description of the concepts and principles involved in the quality control of articles that must be sterile. Any modifications of or variations in sterility test procedures from those described under Sterility Tests  71
71 should be validated in the context of the entire sterility assurance program and are not intended to be methods alternative to those described in that chapter.
should be validated in the context of the entire sterility assurance program and are not intended to be methods alternative to those described in that chapter.
Within the strictest definition of sterility, a specimen would be deemed sterile only when there is complete absence of viable microorganisms from it. However, this absolute definition cannot currently be applied to an entire lot of finished compendial articles because of limitations in testing. The sterility of a lot purported to be sterile is therefore defined in probabilistic terms, where the likelihood of a contaminated unit or article is acceptably remote. Such a state of sterility assurance can be established only through the use of validated sterilization processes or aseptic processing, if any, under appropriate current good manufacturing practice, and not by reliance solely on sterility testing. The basic principles for validation and certification of a sterilizing process are enumerated as follows:
- Establish that the process equipment has the capability of operating within the required parameters.
- Demonstrate that the critical control equipment and instrumentation are capable of operating within the prescribed parameters for the process equipment.
- Perform replicate cycles representing the required operational range of the equipment and employing actual or simulated product. Demonstrate that the processes have been carried out within the prescribed protocol limits and, finally, that the probability of microbial survival in the replicate processes completed is not greater than the prescribed limits.
- Monitor the validated process during routine operation. Periodically as needed, requalify and recertify the equipment.
- Complete the protocols, and document steps (1) through (4) above.
The principles and implementation of a program to validate an aseptic processing procedure are substantially more extensive than the validation of a sterilization process. In aseptic processing, the components of the final dosage form are sterilized separately and the finished article is assembled in an aseptic manner.
Proper validation of the sterilization process or the aseptic process requires a high level of knowledge of the field of sterilization and clean room technology. In order to comply with currently acceptable and achievable limits in sterilization parameters, it is necessary to employ appropriate instrumentation and equipment to control the critical parameters such as temperature, time, pressure, humidity, sterilizing gas concentration, and/or absorbed radiation. An important aspect of the validation program in many sterilization procedures involves the employment of biological indicators (see Biological Indicators 1035). The validated and certified process should be revalidated periodically; however, the revalidation program need not necessarily be as extensive as the original program.
A typical validation program, as outlined below, is one designed for the steam autoclave, but several of these principles may be applicable to the other sterilization procedures discussed in this informational chapter. The program comprises several stages.
The installation qualification stage is intended to establish that controls and other instrumentation are properly designed and calibrated. Documentation should be on file demonstrating the quality of the required utilities such as steam, water, and air. The operational qualification stage is intended to confirm that the empty chamber functions within the parameters of temperature at key chamber locations prescribed in the protocol. It is usually appropriate to develop heat profile records, i.e., simultaneous temperatures in the chamber employing multiple temperature-sensing devices. A typical acceptable range of temperature in the empty chamber is ±1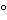 when the chamber temperature is not less than 121. The confirmatory stage of the validation program is the actual sterilization of materials or articles. This determination requires the employment of temperature-sensing devices inserted into samples of the articles, as well as samples of the articles to which appropriate concentrations of suitable test microorganisms (biological indicators) have been added in operationally fully loaded autoclave configurations. The effectiveness of moist heat penetration into the actual articles and the time of the exposure are the two main factors that determine the lethality of the sterilization process. The final stage of the validation program requires the documentation of the supporting data developed in executing the program.
when the chamber temperature is not less than 121. The confirmatory stage of the validation program is the actual sterilization of materials or articles. This determination requires the employment of temperature-sensing devices inserted into samples of the articles, as well as samples of the articles to which appropriate concentrations of suitable test microorganisms (biological indicators) have been added in operationally fully loaded autoclave configurations. The effectiveness of moist heat penetration into the actual articles and the time of the exposure are the two main factors that determine the lethality of the sterilization process. The final stage of the validation program requires the documentation of the supporting data developed in executing the program.
It is generally accepted that terminally sterilized injectable articles or critical devices purporting to be sterile, when sterilized, attain a 10–6 microbial survivor probability, i.e., assurance of less than or equal to 1 chance in 1 million that viable microorganisms are present in the sterilized article or dosage form. With heat-stable articles, the approach often is to exceed the critical time necessary to achieve the 10–6 microbial survivor probability (overkill) of presterilization bioburden that is considerably greater in population (typically 106) and resistance (typically D121 is equal to or greater than 1.0 minute) than the natural presterilization bioburden. However, with an article where extensive heat exposure may have a damaging effect, it will not be feasible to employ an overkill approach. In this latter instance, the development of the sterilization cycle depends heavily on knowledge of the population and resistance microbial burden of the product, based on examination, over a suitable time period, of a substantial number of lots of the presterilized product.
The D value is the time (in minutes) required to reduce the microbial population by 90% or 1 log cycle (i.e., to a surviving fraction of 1/10), at a specific lethal condition, such as, temperature. Therefore, where the D value of a BI preparation of, for example, Geo bacillus stearothermophilus spores is 1.5 minutes under the process conditions defined, e.g., at 121, if it is treated for 12 minutes under the same conditions, it can be stated that the lethality input is 8D. The effect of applying this input to the product would depend on the initial microbial burden. Assuming that its resistance to sterilization is equivalent to that of the BI, if the microbial burden of the product in question is 102 microorganisms, a lethality input of 2D yields a microbial burden of 1 (100 theoretical), and a further 6D yields a calculated microbial survivor probability of 10–6. (Under the same conditions, a lethality input of 12D may be used in a typical “overkill” approach.) Generally, the survivor probability achieved for the article under the validated sterilization cycle is not completely correlated with what may occur with the BI. For valid use, therefore, it is essential that the resistance of the BI be greater than that of the natural microbial burden of the article sterilized. It is then appropriate to make a worst-case assumption and treat the microbial burden as though its heat resistance were equivalent to that of the BI, although it is not likely that the most resistant of a typical microbial burden isolates will demonstrate a heat resistance of the magnitude shown by this species, frequently employed as a BI for steam sterilization. In the above example, a 12-minute cycle is considered adequate for sterilization if the product had a microbial burden of 102 microorganisms. However, if the indicator originally had 106 microorganisms content, actually a 10–2 probability of survival could be expected; i.e., 1 in 100 BIs may yield positive results. This type of situation may be avoided by selection of the appropriate BI. Alternatively, high content indicators may be used on the basis of a predetermined acceptable count reduction.
The D value for the Geo bacillus stearothermophilus preparation determined or verified for these conditions should be reestablished when a specific program of validation is changed. Determination of survival curves (see Biological Indicators 1035), or what has been called the fractional cycle approach, may be employed to determine the D value of the biological indicator preferred for the specific sterilization procedure. The fractional cycle approach may also be used to evaluate the resistance of the microbial burden. Fractional cycles are studied either for microbial count-reduction or for fraction negative achievement. These numbers may be used to determine the lethality of the process under production conditions. The data can be used in qualified production equipment to establish appropriate sterilization cycles. A suitable biological indicator such as the Geo bacillus stearothermophilus preparation may be employed also during routine sterilization. Any microbial burden-based sterilization process requires adequate surveillance of the microbial resistance of the article to detect any changes, in addition to periodic surveillance of other attributes.
METHODS OF STERILIZATION
In this informational chapter, five methods of terminal sterilization, including removal of microorganisms by filtration and guidelines for aseptic processing, are described. Modern technological developments, however, have led to the use of additional procedures. These include blow-molding (at high temperatures), forms of moist heat other than saturated steam and UV irradiation, as well as on-line continuous filling in aseptic processing. The choice of the appropriate process for a given dosage form or component requires a high level of knowledge of sterilization techniques and information concerning any effects of the process on the material being sterilized.1
Steam Sterilization
The process of thermal sterilization employing saturated steam under pressure is carried out in a chamber called an autoclave. It is probably the most widely employed sterilization process. The basic principle of operation is that the air in the sterilizing chamber is displaced by the saturated steam, achieved by employing vents or traps. In order to displace air more effectively from the chamber and from within articles, the sterilization cycle may include air and steam evacuation stages. The design or choice of a cycle for given products or components depends on a number of factors, including the heat lability of the material, knowledge of heat penetration into the articles, and other factors described under the validation program (see above). Apart from that description of sterilization cycle parameters, using a temperature of 121, the F0 concept may be appropriate. The F0, at a particular temperature other than 121, is the time (in minutes) required to provide the lethality equivalent to that provided at 121 for a stated time. Modern autoclaves generally operate with a control system that is significantly more responsive than the steam reduction valve of older units that have been in service for many years. In order for these older units to achieve the precision and level of control of the cycle discussed in this chapter, it may be necessary to upgrade or modify the control equipment and instrumentation on these units. This modification is warranted only if the chamber and steam jacket are intact for continued safe use and if deposits that interfere with heat distribution can be removed.
Dry-Heat Sterilization/Depyrogenation
The process of thermal sterilization of Pharmacopeial articles by dry heat may be carried out by a batch process in an oven designed expressly for that purpose or in a dry-heat tunnel in which glass containers move on a continuous basis through the system. A dry-heat sterilization/depyrogenation system is supplied with heated, HEPA filtered air, distributed uniformly throughout the unit by convection or radiation and employing a blower system with devices for sensing, monitoring, and controlling all critical parameters. A typical acceptable range in temperature in the empty chamber is ±15 when the unit is operating at not less than 250.
In addition to the batch process described above, the continuous-tunnel system usually requires a much higher temperature than cited above for the batch process because of a much shorter dwell time. The continuous process also usually necessitates a rapid cooling stage prior to the aseptic filling operation. In the qualification and validation program, in view of the short dwell time, parameters for uniformity of the temperature, and particularly the dwell time, should be established.
Because depyrogenation is a more rigorous challenge for dry-heat processing systems than biological indicator inactivation, it is generally not necessary to include BIs when validating dry-heat processes if validation of depyrogenation is demonstrated. A 3 log cycle reduction or greater is a suitable acceptance criterion for depyrogenation and when successfully demonstrated will ensure not only adequate depyrogenation of compendial articles but also sterilization. Depyrogenation tests are typically done using articles inoculated with reference standard endotoxin. Articles are then evaluated after exposure for residual levels of endotoxin using Limulus lysate-based assays. For additional information on the endotoxin assay, see Bacterial Endotoxins Test 85.
Gas Sterilization
The choice of gas sterilization as an alternative to heat is frequently made when the material to be sterilized cannot withstand the high temperatures obtained in the steam sterilization or dry-heat sterilization processes. The most commonly employed method of gaseous sterilization is ethylene oxide. Among the disadvantages of ethylene oxide are its highly flammable nature unless mixed with suitable inert gases, its mutagenic properties, and the possibility of toxic residues in treated materials, particularly those containing chloride ions. The sterilization process is generally carried out in a pressure and vacuum-rated chamber designed similarly to a steam autoclave but with the additional features (see below) unique to sterilizers employing this gas. Facilities employing this sterilizing agent should be designed to provide adequate post sterilization degassing, to enable microbial survivor monitoring, and to minimize exposure of operators to the potentially harmful gas.2
Validation of a sterilizing process employing ethylene oxide gas is accomplished along the lines discussed earlier. However, the program is more comprehensive than for the other sterilization procedures, because in addition to temperature, the humidity, vacuum/positive pressure, and ethylene oxide concentration also require appropriate parametric control. An important determination is to demonstrate that all critical process parameters in the chamber are adequate during the entire cycle. Because the sterilization parameters applied to the articles to be sterilized are critical variables, it is frequently advisable to precondition the load to achieve the required moisture content in order to minimize the time of holding at the required temperature before placement of the load in the ethylene oxide chamber. Validation is generally conducted employing product inoculated with appropriate BIs such as spore preparations of Bacillus atrophaeus. For validation they may be used in full chamber loads of product, or simulated product. The monitoring of moisture and gas concentration requires the utilization of sophisticated instrumentation that only knowledgeable and experienced individuals can calibrate, operate, and maintain. BIs may also be employed in monitoring routine runs.
As is indicated elsewhere in this chapter, the BI may be employed in a fraction negative mode to establish the ultimate microbiological survivor probability in designing an ethylene oxide sterilization cycle using inoculated product or inoculated simulated product.
One of the principal limiting factors of the ethylene oxide sterilization process is the limited ability of the gas to diffuse to the innermost product areas that require sterilization. Package design and chamber loading patterns therefore must be determined to allow for necessary gas penetration. The reader is referred to ISO 11135 for a complete description of process development, validation, and routine control of ethylene oxide sterilization processes.
Sterilization by Ionizing Radiation
The rapid proliferation of medical devices unable to withstand heat sterilization and the concerns about the safety of ethylene oxide have resulted in increasing applications of radiation sterilization. This method may also be applicable to active pharmaceutical ingredients and final dosage forms. The advantages of sterilization by irradiation include low chemical reactivity, low measurable residues, and the fact that there are fewer variables to control. In fact, radiation sterilization is unique in that the basis of control is essentially that of the absorbed radiation dose, which can be precisely measured. Dose-setting and dose-substantiation procedures are typically used to validate the radiation dose required to achieve a sterility assurance level. Irradiation causes only a minimal temperature rise but can affect certain grades and types of plastics and glass.
The two types of ionizing radiation in use are radioisotope decay (gamma radiation) and electron-beam radiation. In either case the radiation dose established to yield the required degree of sterility assurance should be such that, within the range of minimum and maximum doses set, the properties of the article being sterilized are acceptable. The reader is referred to ISO 11137-1, - 2, and -3 for a complete description of process development, validation, and routine control of ionizing radiation processes.
Sterilization by Filtration
The sterilization of fluids by filtration is a separative process and differs from the other methods of sterilization that rely on destructive mechanisms. Filtration through microbial retentive materials is frequently employed for the sterilization of heat-labile solutions by physical removal of the contained microorganisms. A filter assembly generally consists of a porous matrix integrated with or clamped into a housing. The effectiveness of a filter medium depends upon the pore size of the porous material, the prefiltration bioburden, and may depend upon adsorption of bacteria on or in the filter matrix or upon a sieving mechanism. There is some evidence to indicate that sieving is the more important component of the mechanism. While fiber-shedding filters are to be avoided unless no alternative filtration procedures are possible, it should be noted that in accordance with 21CFR 211.72, the use of asbestos-containing filters is prohibited. Where a fiber-shedding filter is required, it is obligatory that the process include a nonfiber-shedding filter introduced downstream or subsequent to the initial filtration step.
Filter Rating—
The pore sizes of filter membranes are rated by a nominal rating that reflects the capability of the filter membrane to retain microorganisms of size represented by specified strains, not by determination of an average pore size and statement of distribution of sizes. Sterilizing filters cannot be narrowly defined because, depending upon the bioburden present in the fluid stream, different filters may be considered effective for sterilization. Currently a sterilizing filter can be defined as, “a filter that, when appropriately validated, will remove all microorganisms from a fluid stream, producing a sterile effluent”. The nominal ratings of sterilizing filters based on microbial retention properties differ when rating is done by other means, e.g., by retention of latex spheres of various diameters. It is the user's responsibility to select a filter of correct rating for the particular purpose, depending on the nature of the product (especially considering its potential bioburden) to be filtered. It is not feasible to repeat the tests of filtration capacity in the user's establishment. Microbial challenge tests are preferably performed under a manufacturer's conditions on each lot of manufactured filter membranes.
The user must determine whether filtration parameters employed in manufacturing will significantly influence microbial retention efficiency. Some of the other important concerns in the validation of the filtration process include product compatibility, sorption of drug, preservative or other additives, and initial effluent endotoxin content.
Because the effectiveness of the filtration process is also influenced by the microbial burden of the solution to be filtered, determining the microbiological quality of solutions prior to filtration is an important aspect of the validation of the filtration process, in addition to establishing the other parameters of the filtration procedure, such as pressures, flow rates, and filter unit characteristics. Hence, another method of describing filter-retaining capability is the use of the log reduction value (LRV). For instance, a 0.2-µm filter that can retain 107 microorganisms of a specified strain will have an LRV of not less than 7 under the stated conditions.
The housings and filter assemblies that are chosen should first be validated for compatibility and integrity by the user. While it may be possible to mix assemblies and filter membranes produced by different manufacturers, the compatibility of these hybrid assemblies should first be validated. Additionally, there are other tests to be made by the manufacturer of the membrane filter, which are not usually repeated by the user. These include microbiological challenge tests. Results of these tests on each lot of manufactured filter membranes should be obtained from the manufacturer by users for their records.
Filtration for sterilization purposes is usually carried out with assemblies having membranes of nominal pore size rating of 0.2 µm or less. A membrane filter assembly must be tested for initial integrity prior to use, provided that such test does not impair the safety, integrity, and validity of the system, and should be tested after the filtration process is completed to demonstrate that the filter assembly maintained its integrity throughout the entire filtration procedure. Typical use tests are the bubble point test, the diffusive airflow test, the pressure hold test, and the forward flow test. These tests should be correlated with microorganism retention.
Unidirectional Aseptic Processing
Although there is general agreement that sterilization of the final filled container as a dosage form or final packaged device is the preferred process for ensuring the minimal risk of microbial contamination in a lot, there is a substantial class of products that are not terminally sterilized but are prepared by a series of aseptic steps. These are designed to prevent the introduction of viable microorganisms into components, where sterile, or once an intermediate process has rendered the bulk product or its components free from viable microorganisms. The fundamental difference between aseptically produced sterile products and terminally sterilized products is the presence of a step that can be validated, whereby the final package is subjected to conditions shown to kill viable contaminants. Consequently, an aseptically filled product labeled as sterile must use a system of risk assessments to establish that an acceptable level of sterility assurance has been achieved. Current technology cannot provide an adequate safety assessment based on individual unit testing. In currently used methods of environmental monitoring, process simulations have not been shown to correlate directly with contaminated finished products. Finished product destructive testing (sterility tests) can only examine a very small percentage of a lot and are thus only capable of detecting grossly contaminated lots. This section provides a review of the principles involved in producing aseptically processed products with a minimal risk of microbial contamination in the finished lot of final dosage forms.
A product defined as aseptically processed is likely to consist of components that have been sterilized by one of the processes described earlier in this chapter. For example, the bulk product, if a filterable liquid, may have been sterilized by filtration. The final empty container components would probably be sterilized by heat, dry heat being employed for glass vials and an autoclave being employed for rubber closures. The areas of critical concern are the immediate microbial environment where these presterilized components are exposed during assembly to produce the finished dosage form and the aseptic filling operation.
The requirements for a properly designed, validated, and maintained filling or other aseptic processing facility are mainly directed to (1) an air environment that is suitably controlled with respect to viable and nonviable particulates, of a proper design to permit effective maintenance of air supply units, and (2) the provision of trained operating personnel who are adequately equipped and gowned. The desired environment may be achieved through the high level of air filtration technology now available, which contributes to the delivery of air of the requisite microbiological quality.3 The facilities include both primary (in the vicinity of the exposed article) and secondary (where the aseptic processing is carried out) barrier systems.
For a properly designed aseptic processing facility or aseptic filling area, consideration should be given to such features as nonporous and smooth surfaces, including walls and ceilings that can withstand routine decontamination; gowning rooms with adequate space for personnel and storage of sterile garments; adequate separation of preparatory rooms for personnel from final aseptic processing rooms, with the availability, if necessary, of devices such as airlocks and air showers; proper pressure differentials between rooms, the most positive pressure being in the aseptic processing rooms or areas; the employment of unidirectional airflow in the immediate vicinity of exposed product or components, and filtered air exposure thereto, with adequate air change frequency; appropriate humidity and temperature environmental controls; and a documented sanitization program. Proper training of personnel in hygienic and gowning techniques should be undertaken so that, for example, gowns, gloves, and other body coverings substantially cover exposed skin surfaces.
Certification and validation of the aseptic process and facility are achieved by establishing the efficiency of the filtration systems, by employing microbiological environmental monitoring procedures, and by processing of sterile culture medium as simulated product.
Monitoring of the aseptic facility should include periodic HEPA filter evaluation and testing, as well as routine particulate and microbiological environmental monitoring. Periodic media-fill or process-simulation testing should also be performed.
STERILITY TESTING OF LOTS
It should be recognized that the referee sterility test might not detect microbial contamination if present in only a small percentage of the finished articles in the lot because the specified number of units to be taken imposes a significant statistical limitation on the utility of the test results. This inherent limitation, however, has to be accepted, because current knowledge offers no nondestructive alternatives for ascertaining the microbiological quality of every finished article in the lot, and it is not a feasible option to increase the number of specimens significantly. For information regarding the conduct of the sterility test please see Sterility Tests 71.
1
Documents addressing the development and validation of sterilization cycles and related topics include, by the Parenteral Drug Association, Inc. (PDA), Validation of Moist Heat Sterilization Processes: Cycle Design, Development, Qualification and Ongoing Control (Technical Report No. 1); Process Simulation for Aseptically Filled Products (Technical Report No. 22); Sterilizing Filtration of Liquids (Technical Report No. 26); and Validation of Dry Heat Processes Used for Sterilization and Depyrogenation (Technical Monograph No. 3); and by the Pharmaceutical Manufacturers Association (PMA), Validation of Sterilization of Large-Volume Parenterals—Current Concepts (Science and Technology Publication No. 25). Other technical publications include Health Industry Manufacturers Association (HIMA), Validation of Sterilization Systems (Report No. 78-4.1); Sterilization Cycle Development (Report No. 78-4.2); Industrial Sterility: Medical Device Standards and Guidelines (Document #9, Vol. 1); and Operator Training . . . for Ethylene Oxide Sterilization, for Steam Sterilization Equipment, for Dry Heat Sterilization Equipment, and for Radiation Sterilization Equipment (Report Nos. 78-4.5 through 4.8). Recommended practice guidelines published by the Association for the Advancement of Medical Instrumentation (AAMI) include Guideline for Industrial Ethylene Oxide Sterilization of Medical Devices—Process Design, Validation, Routine Sterilization (No. OPEO-12/81) and Process Control Guidelines for the Radiation Sterilization of Medical Devices (No. RS-P 10/82). Additional radiation sterilization content can be found in ISO 11137—Sterilization of Health Care Products—Requirements for Validation and Routine Control—Radiation Sterilization. These more detailed publications should be consulted for more extensive treatment of the principles and procedures described in this chapter.
2
See Ethylene Oxide, Encyclopedia of Industrial Chemical Analysis, 1971, 12, 317–340, John Wiley & Sons, Inc., and Use of Ethylene Oxide as a Sterilant in Medical Facilities, NIOSH Special Occupational Hazard Review with Control Recommendations, August 1977, U.S. Department of Health and Human Services, Public Health Service, Centers for Disease Control and Prevention, National Institute for Occupational Safety and Health, Division of Criteria Documentation and Standards Development, Priorities and Research Analysis Branch, Rockville, MD.
3
Available published standards for such controlled work areas include the following: (1) ISO 14464 1-7 Cleanrooms and Associated Controlled Environments. (2) NASA Standard for Clean Room and Work Stations for Microbially Controlled Environment, publication NHB5340.2, Aug. 1967. (3) Contamination Control of Aerospace Facilities, U.S. Air Force, T.O. 00-25-203, 1 Dec. 1972, change 1-1, Oct. 1974.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Radhakrishna S Tirumalai, Ph.D.
Principal Scientific Liaison (301) 816-8339 |
(GCM2010) General Chapters - Microbiology |
USP38–NF33 Page 1427
Pharmacopeial Forum: Volume No. 35(4) Page 952