



Change to read:
Purpose
The Dissolution Procedure: Development and Validation  1092
1092 provides a comprehensive approach covering items to consider for developing and validating dissolution procedures and the accompanying analytical procedures. It addresses the use of automation throughout the test and provides guidance and criteria for validation. It also addresses the treatment of the data generated and the interpretation of acceptance criteria for immediate- and modified-release solid oral dosage forms.
provides a comprehensive approach covering items to consider for developing and validating dissolution procedures and the accompanying analytical procedures. It addresses the use of automation throughout the test and provides guidance and criteria for validation. It also addresses the treatment of the data generated and the interpretation of acceptance criteria for immediate- and modified-release solid oral dosage forms.
Scope
Chapter1092 addresses the development and validation of dissolution procedures, with a focus on solid oral dosage forms. Many of the concepts presented, however, may be applicable to other dosage forms and routes of administration. General recommendations are given with the understanding that modifications of the apparatus and procedures as given in USP general chapters need to be justified.
The organization of 1092 follows the sequence of actions often performed in the development and validation of a dissolution test. The sections appear in the following sequence.
-
PRELIMINARY ASSESSMENT (FOR EARLY STAGES OF PRODUCT DEVELOPMENT/DISSOLUTION METHOD DEVELOPMENT)
1.1 Performing Filter Compatibility
1.2 Determining Solubility and Stability of Drug Substance in Various Media
1.3 Choosing a Medium and Volume
1.4 Choosing an Apparatus -
METHOD DEVELOPMENT
2.1 Deaeration
2.2 Sinkers
2.3 Agitation
2.4 Study Design
2.4.1 Time Points
2.4.2 Observations
2.4.3 Sampling
2.4.4 Cleaning
2.5 Data Handling
2.6 Dissolution Procedure Assessment -
ANALYTICAL FINISH
3.1 Sample Processing
3.2 Filters
3.3 Centrifugation
3.4 Analytical Procedure
3.5 Spectrophotometric Analysis
3.6 HPLC -
AUTOMATION
4.1 Medium Preparation
4.2 Sample Introduction and Timing
4.3 Sampling and Filtration
4.4 Cleaning
4.5 Operating Software and Computation of Results
4.6 Common Deviations from the Compendia Procedures That May Require Validation -
VALIDATION
5.1 Specificity/Placebo Interference
5.2 Linearity and Range
5.3 Accuracy/Recovery
5.4 Precision
5.4.1 Repeatability of Analysis
5.4.2 Intermediate Precision/Ruggedness
5.4.3 Reproducibility
5.5 Robustness
5.6 Stability of Standard and Sample Solutions
5.7 Considerations for Automation -
ACCEPTANCE CRITERIA
6.1 Immediate-Release Dosage Forms
6.2 Delayed-Release Dosage Forms
6.3 Extended-Release Dosage Forms
6.4 Multiple Dissolution Tests
6.5 Interpretation of Dissolution Results
6.5.1 Immediate-Release Dosage Forms
6.5.2 Delayed-Release Dosage Forms
6.5.3 Extended-Release Dosage Forms -
REFERENCES
1.1Performing Filter Compatibility
Filtration is a key sample-preparation step in achieving accurate test results. The purpose of filtration is to remove undissolved drug and excipients from the withdrawn solution. If not removed from the sample solution, particles of the drug will continue to dissolve and can bias the results. Therefore, filtering the dissolution samples is usually necessary and should be done immediately if the filter is not positioned on the cannula.
Filtration also removes insoluble excipients that may otherwise interfere with the analytical finish. Selection of the proper filter material is important and should be accomplished, and experimentally justified, early in the development of the dissolution procedure. Important characteristics to consider when choosing a filter material are type, filter size, and pore size. The filter that is selected based on evaluation during the early stages of dissolution procedure development may need to be reconsidered at a later time point. Requalification has to be considered after a change in composition of the drug product or changes in the quality of the ingredients (e.g. particle size of microcrystalline cellulose).
Examples of filters used in dissolution testing can be cannula filters, filter disks or frits, filter tips, or syringe filters. The filter material has to be compatible with the media and the drug. Common pore sizes range from 0.20 to 70 µm, however, filters of other pore sizes can be used as needed. If the drug substance particle size is very small (e.g., micronized or nanoparticles), it can be challenging to find a filter pore size that excludes these small particles.
Adsorption of the drug(s) by the filter may occur and needs to be evaluated. Filter materials will interact with dissolution media to affect the recovery of the individual solutes and must be considered on a case-by-case basis. Different filter materials exhibit different drug-binding properties. Percentage of drug loss from the filtrate due to binding may be dependent on the drug concentration. Therefore the adsorptive interference should be evaluated on sample solutions at different concentrations bracketing the expected concentration range. Where the drug adsorption is saturable, discarding an initial volume of filtrate may allow the collection of a subsequent solution that approaches the original solution concentration. Alternative filter materials that minimize adsorptive interference can usually be found. Prewetting of the filter with the medium may be necessary. In addition, it is important that leachables from the filter do not interfere with the analytical procedure. This can be evaluated by analyzing the filtered dissolution medium and comparing it with the unfiltered medium.
The filter size should be based on the volume to be withdrawn and the amount of particles to be separated. Use of the correct filter dimensions will improve throughput and recovery, and also reduce clogging. Use of a large filter for small-volume filtration can lead to loss of sample through hold-up volume, whereas filtration through small filter sizes needs higher pressures and longer times, and the filters can clog quickly.
Filters used for USP Apparatus 4 need special attention because they are integrated in the flow-through process. Undissolved particles may deposit on the filters, creating resistance to the flow.
In the case of automated systems, selection of the filter with regard to material and pore size can be done in a similar manner to manual filtration. Flow rate through the filter and clogging may be critical for filters used in automated systems. Experimental verification that a filter is appropriate may be accomplished by comparing the responses for filtered and unfiltered standard and sample solutions. This is done by first preparing a suitable standard solution and a sample solution. For example, prepare a typical dissolution sample in a beaker and stir vigorously with a magnetic stirrer to dissolve the drug load completely. For standard solutions, compare the results for filtered solutions (after discarding the appropriate volume) to those for the unfiltered solutions. For sample solutions, compare the results for filtered solutions (after discarding the appropriate volume) to those for centrifuged, unfiltered solutions.
1.2 Determining Solubility and Stability of Drug Substance in Various Media
Physical and chemical characteristics of the drug substance need to be determined as part of the process of selecting the proper dissolution medium. When deciding the composition of the medium for dissolution testing, it is important to evaluate the influence of buffers, pH, and if needed, different surfactants on the solubility and stability of the drug substance. Solubility of the drug substance is usually evaluated by determining the saturation concentration of the drug in different media at 37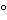 using the shake-flask solubility method (equilibrium solubility). To level out potential ion effects between the drug and the buffers used in the media, mixtures of hydrochloric acid and sodium hydroxide are used to perform solubility investigations; this is in addition to the typical buffer solutions. In certain cases, it may be necessary to evaluate the solubility of the drug at temperatures other than 37 (i.e., 25). The pH of the clear supernatant should be checked to determine whether the pH changes during the solubility test. Alternative approaches for solubility determination may also be used.
using the shake-flask solubility method (equilibrium solubility). To level out potential ion effects between the drug and the buffers used in the media, mixtures of hydrochloric acid and sodium hydroxide are used to perform solubility investigations; this is in addition to the typical buffer solutions. In certain cases, it may be necessary to evaluate the solubility of the drug at temperatures other than 37 (i.e., 25). The pH of the clear supernatant should be checked to determine whether the pH changes during the solubility test. Alternative approaches for solubility determination may also be used.
Typical media for dissolution may include the following (not listed in order of preference): diluted hydrochloric acid, buffers (phosphate or acetate) in the physiologic pH range of 1.2–7.5, simulated gastric or intestinal fluid (with or without enzymes), and water. For some drugs, incompatibility of the drug with certain buffers or salts may influence the choice of buffer. The molarity of the buffers and acids used can influence the solubilizing effect, and this factor may be evaluated.
Aqueous solutions (acidic or buffer solutions) may contain a percentage of a surfactant [e.g., sodium dodecyl sulfate (SDS), polysorbate, or lauryldimethylamine oxide] to enhance the solubility of the drug. The surfactants selected for the solubility investigations should cover all common surfactant types, i.e., anionic, nonionic, and cationic. When a suitable surfactant has been identified, different concentrations of that surfactant should be investigated to identify the lowest concentration needed to achieve sink conditions. Typically, the surfactant concentration is above its critical micellar concentration (CMC). Table 1 shows a list of some of the surfactants used in dissolution media. Approximate CMC values are provided with references when available. The list is not comprehensive and is not intended to exclude surfactants that are not listed. Other substances, such as hydroxypropyl 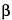 -cyclodextrin, have been used as dissolution media additives to enhance dissolution of poorly soluble compounds. The U.S. Food and Drug Administration (FDA) maintains a database of dissolution methods, including information on dissolution media that have been used (1). Typically, the amount of surfactant added is sufficient to achieve sink conditions in the desired volume of dissolution medium.
-cyclodextrin, have been used as dissolution media additives to enhance dissolution of poorly soluble compounds. The U.S. Food and Drug Administration (FDA) maintains a database of dissolution methods, including information on dissolution media that have been used (1). Typically, the amount of surfactant added is sufficient to achieve sink conditions in the desired volume of dissolution medium.
It is important to control the grade and purity of surfactants because use of different grades could affect the solubility of the drug. For example, SDS is available in both a technical grade and a high-purity grade. Obtaining polysorbate 80 from different sources can affect its suitability when performing high-performance liquid chromatography (HPLC) analysis.
There may be effects of counter-ions or pH on the solubility or solution stability of the surfactant solutions. For example, a precipitate forms when the potassium salt for the phosphate buffer is used at a concentration of 0.5 M in combination with SDS. This can be avoided by using the sodium phosphate salt when preparing media with SDS.
Table 1. Commonly Used Surfactants with Critical Micelle Concentrations
| Surfactant
|
CMC (% wt/volume) |
Reference |
|
|---|---|---|---|
| Anionic |
Sodium dodecyl sulfate (SDS), Sodium lauryl sulfate (SLS) |
0.18%–0.23% |
(2–4)
|
| Taurocholic acid sodium salt |
0.2% |
(3)
|
|
| Cholic acid sodium salt |
0.16% |
(3)
|
|
| Desoxycholic acid sodium salt |
0.12% |
(3)
|
|
| Cationic |
Cetyltrimethyl ammonium bromide (CTAB, Hexadecyltrimethylammonium bromide) |
0.033%–0.036% (0.92–1.0 mM) |
(5,6)
|
| Benzethonium chloride (Hyamine 1622) |
0.18% (4 mM) |
(2)
|
|
| Nonionic |
Polysorbate 20 (Polyoxyethylene (20) sorbitan monolaurate, Tween 20) |
0.07%–0.09% |
(3,7)
|
| Polysorbate 80 (Polyoxyethylene (20) sorbitan monooleate, Tween 80) |
0.02%–0.08% |
(3,7)
|
|
| Caprylocaproyl polyoxyl-8 glycerides (Labrasol) |
0.01% |
(4)
|
|
| Polyoxyl 35 castor oil (Cremophor EL) |
0.02% |
(8)
|
|
| Polyoxyethylene 23 lauryl ether (Brij 35) |
0.013% |
(9)
|
|
| Octoxinol (Triton X-100) |
0.01%–0.03% |
(3,10)
|
|
| Zwitterion |
Lauryldimethylamine N-oxide (LDAO) |
0.023% |
(11)
|
Routinely, the dissolution medium is buffered; however, the use of purified water as the dissolution medium is suitable for products with a dissolution behavior independent of the pH of the medium. There are several reasons why purified water may not be preferred. The water quality can vary depending on its source, and the pH of the water is not as strictly controlled as the pH of buffer solutions. Additionally, the pH can vary from day to day and can also change during the run, depending on the drug substance and excipients. Use of an aqueous–organic solvent mixture as a dissolution medium is discouraged; however, with proper justification this type of medium may be acceptable.
Investigations of the stability of the drug substance should be carried out, when needed, in the selected dissolution medium with excipients present, at 37. This elevated temperature has the potential to decrease solution stability (degradation). Stability should allow for sufficient time to complete or repeat the analytical procedure. Physical stability may be of concern when precipitation occurs because of lower solubility at room or refrigerated temperature.
1.3 Choosing a Medium and Volume
When developing a dissolution procedure, one goal is to have sink conditions, which are defined as having a volume of medium at least three times the volume required to form a saturated solution of drug substance. When sink conditions are present, it is more likely that dissolution results will reflect the properties of the dosage form. A medium that fails to provide sink conditions may be acceptable if it is appropriately justified. The composition and volume of dissolution medium are guided by the solubility investigations. For example, the choice and concentration of a surfactant need to be justified from the solubility data and the dissolution profiles.
The use of enzymes in the dissolution medium is permitted, in accordance with Dissolution 711, when dissolution failures occur as a result of cross-linking with gelatin capsules or gelatin-coated products. A discussion of the phenomenon of cross-linking and method development using enzymes can be found in Capsules–Dissolution Testing and Related Quality Attributes 1094. Validation should be performed with the method using enzymes according to section 5. Validation.
Another option is to use media that follow more closely the composition of fluids in the stomach and intestinal tract. These media may contain physiological surface-active ingredients, such as taurocholates. The media also may contain emulsifiers (lecithin) and components such as saline solution that increase osmolality. Also, the ionic strength or molarity of the buffer solutions may be manipulated. The media are designed to represent the fed and fasted state in the stomach and small intestine. These media may be very useful in modeling in vivo dissolution behavior of immediate-release (IR) dosage forms, in particular those containing lipophilic drug substances, and may help in understanding the dissolution kinetics of the product related to the physiological make-up of the digestive fluids. Results of successful modeling of dissolution kinetics have been published, mainly for IR products. In the case of extended-release dosage forms with reduced effect of the drug substance on dissolution behavior, the use of such media needs to be evaluated differently. In vitro performance testing does not necessarily require media modeling the fasted and postprandial states (12,13).
An acid stage is part of the testing of delayed-release products by Method A or Method B in 711. For drugs with acid solubility less than 10% of the label claim or drugs that degrade in acid the usefulness of the acid stage in detecting a coating failure is compromised. This would be handled on a case-by-case basis. Possible resolutions include the addition of surfactant to the acid stage, or adjustment of the specifications.
During selection of the dissolution medium, care should be taken to ensure that the drug substance is suitably stable throughout the analysis. In some cases, antioxidants such as ascorbic acid may be used in the dissolution medium to stabilize the drug. There are occasions where such actions are not sufficient. For compounds that rapidly degrade to form a stable degradant, monitoring the degradant alone or in combination with a drug substance may be more suitable than analyzing only the drug substance. In situ spectroscopic techniques tend to be less affected by degradation when compared with HPLC analysis (including UHPLC and other liquid chromatographic approaches).
For compendial Apparatus 1 (basket) and Apparatus 2 (paddle), the volume of the dissolution medium can vary from 500 to 1000 mL. Usually, the volume needed for the dissolution test can be determined in order to maintain sink conditions. In some cases, the volume can be increased to between 2 and 4 L, using larger vessels and depending on the concentration and sink conditions of the drug; justification for this approach is expected. In practice, the volume of the dissolution medium is usually maintained within the compendial range given above. Alternatively, it may be preferable to switch to other compendial apparatus, such as a reciprocating cylinder (Apparatus 3), reciprocating holder (Apparatus 7), or flow-through cell (Apparatus 4). Certain applications may require low volumes of dissolution media (e.g., 100–200 mL) when the use of a paddle or basket is preferred. In these cases, an alternative, noncompendial apparatus (e.g., small-volume apparatus) may be used.
1.4 Choosing an Apparatus
The choice of apparatus is based on knowledge of the formulation design and the practical aspects of dosage form performance in the in vitro test system. In general, a compendial apparatus should be selected.
For solid oral dosage forms, Apparatus 1 and Apparatus 2 are used most frequently. When Apparatus 1 or Apparatus 2 is not appropriate, another official apparatus may be used. Apparatus 3 (reciprocating cylinder) has been found especially useful for chewable tablets, soft gelatin capsules, delayed-release dosage forms, and nondisintegrating-type products, such as coated beads. Apparatus 4 (flow-through cell) may offer advantages for modified-release dosage forms and immediate-release dosage forms that contain active ingredients with limited solubility. In addition, Apparatus 4 may have utility for multiple dosage form types such as soft gelatin capsules, beaded products, suppositories, or depot dosage forms, as well as suspension-type extended-release dosage forms. Apparatus 5 (paddle over disk) and Apparatus 6 (rotating cylinder) are useful for evaluating and testing transdermal dosage forms. Apparatus 7 (reciprocating holder) has application to non-disintegrating, oral modified-release dosage forms, stents, and implants, as well as transdermal dosage forms. For semisolid dosage forms, the generally used apparatus include the vertical diffusion cell, immersion cell, and flow-through cell apparatus with the insert for topical dosage forms (see Semisolid Drug Products—Performance Tests 1724).
Some changes can be made to the compendial apparatus; for example, a basket mesh size other than the typical 40-mesh basket (e.g., 10-, 20-, or 80-mesh) may be used when the need is clearly documented by supporting data. Care must be taken that baskets are uniform and meet the dimensional requirements specified in 711.
A noncompendial apparatus may have some utility with proper justification, qualification, and documentation of superiority over the standard equipment. For example, a small-volume apparatus with mini paddles and baskets may be considered for low-dosage strength products. A rotating bottle or dialysis tubes may have utility for microspheres and implants, peak vessels, and modified flow-through cells for special dosage forms including powders and stents.
Many causes of variability can be found in the formulation and manufacturing process. For example, poor content uniformity, process inconsistencies, excipient interactions or interference, film coating, capsule shell aging, and hardening or softening of the dosage form on stability may be sources of variability and interferences.
2.1 Deaeration
The significance of deaeration of the dissolution medium should be determined because air bubbles can act as a barrier to the dissolution process if present on the dosage unit or basket mesh and can adversely affect the reliability of the test results. Furthermore, bubbles can cause particles to cling to the apparatus and vessel walls. Bubbles on the dosage unit may increase buoyancy, leading to an increase in the dissolution rate, or may decrease the available surface area, leading to a decrease in the dissolution rate. Poorly soluble drugs are most sensitive to interference from air bubbles; therefore, deaeration may be needed when testing these types of products. A deaeration method is described as a footnote in the Procedure section of 711. Typical steps include heating the medium, filtering, and drawing a vacuum for a short period of time. Other methods of deaeration are available and are in routine use throughout the industry. Once a suitable deaeration process is identified, it should be documented as part of the dissolution procedure. The extent of deaeration can be evaluated by measuring the total dissolved gas pressure or by measuring the concentration of dissolved oxygen in water. For example, an oxygen concentration below 6 mg/L has been found effective as a marker for adequate deaeration of water for the Performance Verification Test with USP Prednisone Tablets RS.
Media containing surfactants usually are not deaerated because the process results in excessive foaming, and usually the effect of dissolved air on the dissolution process is mitigated by the reduced surface tension of the medium. Sometimes, deaerating the medium before adding surfactants can be effective.
To determine whether deaeration of the medium is necessary, compare results from dissolution samples run in non-deaerated medium and medium deaerated using a compendial technique, as described above. If no effect of deaeration is detected, this experiment could serve as justification that deaeration is not required in the future. If there is an effect, however, then it is necessary to carefully control this parameter, and it is prudent to characterize the robustness of the deaeration process. The dissolved gas content of deaerated media under atmospheric pressure is unstable and will tend toward saturation. Manipulations of the deaerated medium such as stirring or pouring can increase the rate at which atmospheric gases are redissolved.
2.2 Sinkers
Sinkers are often used to adjust the buoyancy of dosage forms that would otherwise float during testing with Apparatus 2. When sinkers are used, a detailed description of the sinker must be provided in the written procedure. It may be useful to evaluate different sinker types, recognizing that sinkers can significantly influence the dissolution profile of a dosage unit. When transferring the procedure, the same sinkers should be used, or if a different design is used, it should be shown to produce equivalent results. There are several types of commercially available sinkers. In 711, a harmonized sinker is described in Figure 2a.
Table 2. Wire Sinkers Used With Common Capsule Shell Sizes
| Capsule Shell Size
|
Length of Wire (cm) |
Diameter Size (cm) |
Cork Bore Number |
|---|---|---|---|
| #0, elongated | 12 | 0.8 | 4 |
| #1 and #2 | 10 | 0.7 | 3 |
| #3 and #4 | 8 | 0.55 | 2 |
A standard sinker can be made by using the appropriate length of wire and coiling it around a cylinder. For materials, use 316 stainless steel wire, typically 0.032 inch/20 gauge, or other inert material and wind the wire around cylinders of appropriate diameter (e.g., cork borers) for an appropriate number of turns to fit the capsule shell type. Sizes are shown in Table 2. The ends of the coil can be curved to retain the capsule within the sinker when they are immersed. Because the ends of the wire may be rough, they may need to be filed. If the sinker is handmade, the sinker material and construction procedure instructions should be documented (e.g., dimension, design, number of coils); if a commercial sinker is used, the vendor part number should be reported if available.
Although sinkers are typically used to keep the dosage form at the bottom of the vessel, they can also be used to keep dosage forms from sticking to the vessel (e.g., film-coated tablets). The sinker should be appropriate to the dosage form; therefore, the same sinker size may not be suitable for all dosage-form sizes. The sinker should not be too tight around the dosage form because this may restrict interaction with the medium. Conversely, if wrapped too loosely, the dosage form may escape soon after the test begins. The sinker should be small enough that the capsule does not change its orientation within the sinker. Care should be taken when testing capsules that have some cross-linking present, to keep the sticky shell from attaching to the vessel bottom. In this case, the harmonized sinker design provided in Figure 2a of 711 will be advantageous.
2.3 Agitation
For immediate-release capsule or tablet formulations, Apparatus 1 (baskets) at 50–100 rpm or Apparatus 2 (paddles) at 50 or 75 rpm are used commonly. Other agitation speeds are acceptable with appropriate justification. Rates outside 25–150 rpm for both the paddle and the basket are usually not appropriate because of mixing inconsistencies that can be generated by stirring too slow or too fast. Agitation rates between 25 and 50 rpm are generally acceptable for suspensions.
For dosage forms that exhibit coning (mounding) under the paddle at 50 rpm, the coning can be reduced by increasing the paddle speed to 75 rpm, thus reducing the artifact and improving the data. If justified, 100 rpm may be used with Apparatus 2, especially for extended-release products. Decreasing or increasing the apparatus rotation speed may be justified if to achieve an in-vitro–in-vivo correlation (IVIVC) the resulting profiles better reflect in vivo performance, or if the method results in better discrimination without adversely affecting method variability.
Apparatus 3 (reciprocating cylinder) can be used at dip rates ranging from 5 to 30 dips/min. The hydrodynamics are influenced by the cylinder's reciprocating motion and the resulting movement of the sample in the medium. The reciprocating motion of the cylinder and screen may cause foaming if the medium contains surfactants. Addition of an anti-foaming agent such as simethicone or n-octanol may be useful for avoiding foaming from surfactants.
Apparatus 4 (flow-through cell) is described in 711 with standard flow rates of 4, 8, and 16 mL/min. Other flow rates for Apparatus 4 can be used if justified and if within the capacity of the pump to conform with the requirements in 711. Agitation in Apparatus 4 is not only related to the pump speed but can also be affected by cell diameter. At a set flow rate, as measured by volume, the 12-mm cell will develop a greater linear fluid velocity than is achieved in the 22.6-mm cell. Apparatus 4 can be configured with the addition of glass beads in the entry cone of the flow-through cell (packed column) or without glass beads (open column).
The flow characteristics of the flow-through cell are discussed in the scientific literature (15). The placement of the sample in the flow-through cell will influence the flow patterns that occur and thus should be a consideration in the attempt to reduce variability of the results.
2.4 Study Design
Selection of the agitation rate and other study design elements for the dosage form, whether immediate release or modified release, should conform to the requirements and specifications (i.e., apparatus, procedures, and interpretation) given in 711.
2.4.1 time points
For immediate-release dosage forms, the duration of the dissolution procedure is typically 30–60 min; in most cases, a single time point specification is adequate for pharmacopeial purposes. For method development, however, a sufficient number of time points should be selected to adequately characterize the ascending and plateau phases of the dissolution curve. Industrial and regulatory concepts of product comparability and performance may require additional time points, which may also be required for product registration or approval. According to the Biopharmaceutics Classification System referred to in several FDA Guidances, highly soluble, highly permeable drugs formulated into very rapidly dissolving products need not be subjected to a profile comparison if they can be shown to release 85% or more of the drug substance within 15 min. For these types of products, a one-point test or disintegration will suffice. However, most products do not fall into this category. Dissolution profiles of immediate-release products typically show a gradual increase reaching 85%–100% at about 30–45 min. Thus, sufficient dissolution time points are chosen to characterize the performance for most immediate-release products. For some products, including suspensions, useful information may be obtained from earlier points, e.g., 5–10 min. For slower-dissolving products, time points later than 60 min may be useful. Dissolution test times for compendial tests are usually established on the basis of an evaluation of the dissolution profile data.
The f2 similarity factor may not be useful when more than 85% is dissolved at 15 min. If the f2 similarity factor is to be used, multiple time points for the dissolution test are required, with at least two time points with mean percent dissolved (typically for n = 12) below 85% dissolved and only one point above 85% for both products (16). Therefore, the addition of early time points may be useful.
For testing an extended-release dosage form, at least three time points are chosen, to guard against dose dumping, to define the in vitro release profile, and to show that essentially complete release (>80%) of the drug is achieved. Additional sampling times may be useful. Certain IVIVC criteria, such as level B correlation (according to In Vitro and In Vivo Evaluation of Dosage Forms 1088), require the experimental determination of the time to dissolve 100% of the label claim. Selection of the final time points is reflective of the data from the drug release profile that are generated during development. For products containing more than a single active ingredient, determine the drug release for each active ingredient.
Delayed-release dosage forms usually require specifications for at least two time points; therefore, it is important during development to evaluate the entire dissolution profile. In the case of enteric-coated dosage forms, the functionality of the coating is usually proven by challenge in an acid medium, followed by a demonstration of dissolution in a higher-pH medium. Chapter 711 gives a standard buffer medium for that stage of testing but other media may be used if justified. The timing of the acid stage is typically 2 h, and release in the buffer is similar to the timing for immediate-release forms. For delayed-release dosage forms that are not enteric coated, setting of specifications is different. Unlike delayed release, the onset of release is not determined by the experimental design, which is the pH change; multivariate specifications, therefore, may be needed to define time ranges and corresponding percentage ranges.
So-called infinity points can be useful during development studies. To obtain an infinity point, the paddle or basket speed is increased at the end of the run (after the last time point) for a sustained period (typically, 15–60 min), after which time an additional sample is taken. Although there is no requirement for 100% dissolution in the profile, the infinity point can be compared to content uniformity data and may provide useful information about formulation characteristics during initial development or about method bias.
2.4.2 observations
Visual observations and recordings of product dissolution and disintegration behavior are useful because dissolution and disintegration patterns can be indicative of variables in the formulation or manufacturing process. For visual observation, proper lighting (with appropriate consideration of photo-degradation) of the vessel contents and clear visibility in the bath are essential. Documenting observations by drawing sketches and taking photographs or videos can be instructive and helpful for those who are not able to observe the real-time dissolution test. Observations are especially useful during method development and formulation optimization. It is important to record observations of all six vessels to determine if the observation is seen in all six vessels, or just a few. If the test is performed to assist with formulation development, provide any unique observations to the formulator. Examples of typical observations include, but are not limited to, the following:
- Uneven distribution of particles throughout the vessel. This can occur when particles cling to the sides of the vessel, when there is coning or mounding directly under the apparatus (e.g., below the basket or paddle), when particles float at the surface of the medium, when film-coated tablets stick to the vessel, and/or when off-center mounds are formed.
- Air bubbles on the inside of the vessel or on the apparatus or dosage unit. Sheen on the apparatus is also a sign of air bubbles. This observation would typically be made when assessing the need to deaerate the medium.
- Dancing or spinning of the dosage unit, or the dosage unit being hit by the paddle.
- Adhesion of particles to the paddle or the inside of the basket, which may be observed upon removal of the stirring device at the end of the run.
- Pellicles or analogous formations, such as transparent sacs or rubbery, swollen masses surrounding the capsule contents.
- Presence of large floating particles or chunks of the dosage unit, especially at the surface of the media.
- Observation of the disintegration rate (e.g., percentage reduction in size of the dosage unit within a certain time frame).
- Complex disintegration of the coating of modified or enteric-coated products, [e.g., the partial opening and splitting apart (similar to a clamshell) or incomplete opening of the shell], accompanied by the release of air bubbles and excipients.
- Whether the dosage form lands in the vessel center or off-center, and if off-center, whether it sticks there.
- Time required for the complete dissolution of the capsule shell or for tablet disintegration.
Observations also help to document that the proper procedure has been followed, or more importantly, that a deviation has occurred. Examples include the confirmation that a dosage form is actually in the vessel during the test or that more than one dosage form are inadvertently in the same vessel, or that a filter from the autosampler has dropped into the vessel.
2.4.3 sampling
Manual:
For manual sampling, use chemically inert devices (e.g., polymeric or glass syringes, and polymeric or stainless steel cannula), a filter, and/or a filter holder. The sampling site must conform to specifications in 711. When the agitation conditions are very slow, e.g., a 50-rpm basket, care should be taken to sample consistently in the same location in the vessel because there may be a concentration gradient; avoid sampling very close to the shaft or vessel wall. During method development, a decision should be made regarding whether to replace the media after each time point. Replacement is not preferred because the dosage unit may be disturbed during delivery of the media. However, replacement may be necessary if maintaining sink conditions is a challenge. With replacement, the volume used in the calculations remains the same throughout the time points, but there is some drug substance withdrawn with each sample that will need to be accounted for in the calculations.
Metal surfaces may interact with the sample. For example, adsorption onto metal surfaces may occur, or the metal surfaces may release metal ions into aqueous media. The ions can then catalyze degradation reactions, leading to artifacts during the analytical procedures. The surfaces of stirring elements and metal locks of syringes may be sources of interference to accurate sampling.
Autosampling:
Autosampling is discussed in section 4. Automation.
2.4.4 cleaning
Importance is placed on evaluation of the cleaning process between tests. Changes of dissolution medium and/or product necessitate the need for cleaning. Residues on the vessels can affect the results (e.g., adsorbed residues may dissolve and alter subsequent media properties or interfere with the sample analysis), and effective cleaning will return them to a suitable state. Automated systems are discussed in section 4.4 Cleaning.
2.5 Data Handling
Dissolution rates are calculated from the change in drug concentration in the dissolution medium. For procedures in which the volume of medium is fixed, such as for Apparatus 1 and Apparatus 2 testing of immediate-release dosage forms with only one sampling time, the concentration of the sample is multiplied by the medium volume to arrive at the mass of drug dissolved usually expressed as percentage of label claim. When multiple time points are taken, the total amount of drug removed at earlier time points should be assessed and may be part of the calculation of the amount dissolved, if considered important. Similarly, if the medium volume is not fixed, for example when the sample volume is not replaced in testing extended-release products, the change in medium volume must be part of the calculation for successive sampling points. Dissolution tests performed with Apparatus 4 in the closed-loop configuration with in situ detection provide a convenient control of the medium volume. For testing with Apparatus 4 in the open configuration, the test time and flow rate will determine the volume of medium used in the dissolution calculations.
Dissolution results can be evaluated as either cumulative rates or fractional rates. Cumulative rates represent the sum of all drug dissolution that occurs during an interval (Figure 1). Fractional rates are assessed at a specific time point or during a portion of the total test time (Figure 2). Typically, the rate of release will be expressed as either mass or percentage of label claim per unit time. For most compendial dissolution testing, the dissolution rate is expressed as a percentage of the label claim dissolved at the indicated test time.
+'&img=../../images/v38332/m643-fig1.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 1. An example of a plot of dissolution as a cumulative process. Concentration, C, is the amount of drug released per volume of medium, and t represents time. This type of plot is readily observed in constant-volume dissolution systems, such as Apparatus 1 or Apparatus 2, or Apparatus 4 in closed-loop configuration.
+'&img=../../images/v38332/m643-fig2.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 2. An example of a plot of the observed concentration of the sample taken for an interval that is negligibly small in relation to the time of the overall dissolution process. This concentration is propostional to the instantaneous or fractional dissolution rate (dc/dt). This type of plot is readily observed in continuous-flow dissolution systems, such as Apparatus 4 in open-loop configuration.
Cumulative dissolution profiles represent the total amount of drug dissolved from the formulation over time. When cumulative dissolution is measured in a constant-volume system, no correction for the amount lost in sampling needs to be made. If sample is removed from the system, the amount consumed in analysis must be accounted for in the calculation. Recirculated sampling with Apparatus 1 or Apparatus 2, or with Apparatus 4 in the closed-loop configuration (Figure 3), are all examples of systems that will produce cumulative dissolution rates. With Apparatus 4 in the open configuration (Figure 4), cumulative rates accounting for the total amount of drug dissolved across the testing interval are obtained by collecting and analyzing the entire outflow from each individual flow-through cell. With Apparatus 3 (Figure 5), the medium in each tube is sampled at the end of the programmed interval, and the analyzed concentration represents the cumulative dissolution rate during that interval.
+'&img=../../images/v38332/m643-fig3.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 3. Apparatus 4 in the closed-loop configuration.
+'&img=../../images/v38332/m643-fig4.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 4. Apparatus 4 in the open-loop configuration. The sample can be collected in fractions, as shown at the top. The medium can be changed by using successive reservoirs.
+'&img=../../images/v38332/m643-fig5.gif&casNumber=&usp=38&nf=33&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 5. The progression that is possible for one reciprocating cylinder from Apparatus 3. The reciprocating cylinder can move from vessel to vessel. This feature facilitates changing the dissolution medium and testing for different intervals in successive tubes.
Fractional dissolution rates are typically measured for a discrete interval. A series of such rates will produce a step function as the dissolution profile. At any time, the cumulative dissolution rate from this type of profile is the sum of the preceding intervals. This type of profile is represented by Apparatus 3 using multiple tubes or Apparatus 4 in the open-loop configuration where the total outflow is collected and analyzed for successive intervals.
A number of algebraic and numerical methods exist for transforming cumulative and fractional dissolution results. The difference in amount released for successive time points can be calculated, and the average release rate is determined by the formula:
Result = (M2 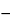 M1)/(t2 t1)
M1)/(t2 t1)
M = mass or percentage of label claim
t = time
As the difference of t2 from t1 is reduced, the average rate can be considered to approach an instantaneous rate. Sampling considerations and physical constraints on measurement of the mass transfer at the medium interface of the dosage form make the measurement of true instantaneous dissolution impractical for routine determination in the laboratory. Fractional dissolution is measured for intervals where the difference between t2 and t1 is small, relative to the total test time. The design of Apparatus 4 in the open configuration permits a direct measurement of the fractional dissolution over small time intervals. For example, if a 4-mL fraction of outflow for Apparatus 4 running 16 mL/min is sampled, either by in situ detection or offline, the amount of drug detected represents the dissolution occurring in a 15-s interval.
Pooled dissolution has been used in a number of monographs. The pooled dissolution procedure produces an average release rate for the units tested by combining equal volumes from each vessel or cell and performing analysis of only the one resulting solution. Because this approach uses only the average release rate for comparison with the acceptance table, the pooled dissolution procedure has been viewed as reducing the amount of data available from the dissolution test and, thus, reducing its value. However, it should be noted that the pooling of equal sample volumes is equivalent, from a calculation standpoint, to determining the arithmetic mean of the individual sample results.
The use of the f2 similarity factor in the comparison of dissolution profiles is discussed in Assessment of Drug Product Performance—Bioavailability, Bioequivalence, and Dissolution 1090.
For the purpose of correlation with in vivo data, parameters of mathematical models are obtained by fitting to dissolution data to establish a continuous functional relationship called IVIVC (see 1088).
2.6 Dissolution Procedure Assessment
The dissolution procedure requires an apparatus, a dissolution medium, and test conditions that together provide a method that is sensitive to changes in critical quality attributes, yet sufficiently rugged and reproducible for day-to-day operation. The method should be able to be transferred between laboratories.
The ideal dissolution procedure will not contribute an unacceptable degree of variability and will provide a profile with adequate points below 85% dissolved. If 85% dissolved occurs before 15 min, then f2 comparisons may not be appropriate.
There are many ways to challenge the sensitivity of the method. One option is to compare dissolution profiles of formulations that are intentionally manufactured with meaningful variations for the most relevant critical manufacturing variable, for example, ±10%–20% change to the ranges of these variables. Similarly, samples that have been stressed may be used to demonstrate sensitivity to changes on stability. This concept may be used to establish the factors that are most significant in their influence on the dissolution rate. These studies can focus on either the dissolution parameters (e.g., media concentration, agitation rate, and deaeration) or the product attributes (e.g., excipient ratios, particle size, compression). The ultimate goal is to understand the release mechanisms and determine whether the dissolution procedure can show change in the critical quality attributes of a drug product.
3.1 Sample Processing
After the samples are withdrawn from the dissolution medium, they may require additional processing to make them suitable for the analytical methodology used to determine the amount released. For example, filtration may be used to remove undissolved particulate matter, or samples may need to be protected from exposure to light or may need refrigerated storage. In addition, samples may have to be diluted to a level that is within the linear range of the method. With analysis by HPLC, dilution of the sample with mobile phase may be necessary to reduce the effect on the separation of injecting dissolution medium. Other types of treatment may be necessary depending on the product formulation, such as the inactivation or elimination of interference caused by components of the formulation by the addition of appropriate reagents. However, separation may not be possible or needed in all cases. In some cases, in situ measurements obtained with methods such as fiber optics or electrochemical determination may be useful.
3.2 Filters
The topic of filtration is discussed in section 1.1 Performing Filter Compatibility.
3.3 Centrifugation
Centrifugation of samples is not preferred, for several reasons: dissolution can continue to occur until the solids are removed, a concentration gradient may form in the supernatant, and energy imparted may lead to increased dissolution of the drug substance particles. Possible exceptions, when centrifugation could be preferred, might include the use with compounds that adsorb onto all common filters, or situations when the potential filter leachables and extractables might interfere in the quantitative step of the dissolution test (e.g., when fluorescence procedures are used in quantitation). Centrifugation may prove useful during method development for evaluating the suitability of the filter material.
3.4 Analytical Procedure
The usual assay for a dissolution sample employs either a spectrophotometric procedure or a liquid chromatographic procedure. Spectrophotometric determination may be direct or may provide the detection for HPLC. Spectrophotometric determination is used often because results can be obtained faster, the analysis is simpler, it is easier to automate, and fewer solvents are needed. The use of direct spectrophotometric determination typically requires confirmation of specificity. HPLC is preferred for a number of reasons such as providing a wide dynamic range that reduces the need to dilute some samples while also providing sensitivity in the analysis of dilute samples, and greater selectivity when excipients or multiple drugs in the formulation present a significant interference. Modern HPLC systems employ autosamplers that provide speed and simplicity advantages comparable to spectrophotometric analysis.
3.5 Spectrophotometric Analysis
Direct spectrophotometric analysis may be performed on samples that are manually introduced to the cuvette. Alternatively, samples may be automatically introduced into the spectrophotometer using autosippers and flow cells. Routine performance checks, cleaning, and maintenance, as described in the standard operating procedures or metrology documents, help to ensure reliable operation of these instruments. Cells with path lengths ranging from 0.02 cm to 1 cm are typically used, and longer path-length cuvettes can be used to increase the range for quantification of dilute samples. Cell alignment and air bubbles could be sources of error. The shorter path-length cells are used to avoid diluting the sample; in all cases, however, acceptable linearity and standard error need to be demonstrated.
The choice of wavelength for the determination should be based on the spectrum of the drug in solution. In some cases, where the drug substance can degrade in the dissolution medium (e.g., dosage forms containing aspirin), it is useful to carry out the measurements at the isosbestic point. Excipients can also have effects, but performing analysis at multiple wavelengths can minimize their effects. The contribution of the absorbance from an excipient at the analytical wavelength can sometimes be determined by ratio from its absorbance at a wavelength where the absorbance of the drug substance is minimal.
Using a validated analytical finish, standard solutions are typically prepared in dissolution media and analyzed at just one concentration, either at 100% of the dosage strength or the selected Q value because linearity of the analytical finish has been established. Prior to validation, dissolution profile analysis, or analysis of products of various strengths, requires using multiple standard solutions covering the expected range of concentration. A typical media blank, standard, and sample may be analyzed in a sequence that brackets the sample with standards and blanks, especially at the beginning and end of the analysis. The standard and sample solutions should both be prepared in the dissolution medium in the linear concentration range and measured at the same wavelength. However, small amounts of an organic solvent may be used in the preparation of the standard, provided that the accuracy criteria can be met during validation.
The absorptivity is calculated by dividing the mean standard absorbance by the concentration, in mg/mL, divided by the cell path length in cm. A rearrangement of the Beer-Lambert expression gives the absorptivity, a, as:
a = A/bc
A = absorbance
b = path length (cm)
c = concentration (mg/mL)
Typical units for absorptivity that are used for dissolution testing are in terms of AU · mL/mg, where AU is absorbance unit. Historical data may be used to provide an acceptable absorptivity range for the analyte (using the appropriate path-length cell). This value may be useful in troubleshooting aberrant data.
Fiber optics as a sampling and determinative method, with proper validation, are an option.
3.6 HPLC
For HPLC analysis, the effect on the chromatogram of peaks resulting from injection of dissolution media require enumeration. A large solvent disturbance may affect accuracy and precision of response if it is poorly resolved from the peak of interest. This is even more important if large injector volumes (>100 µL) are needed. System suitability tests may evaluate peak shape; separation of the main peak from solvent disturbance and from closely eluting peaks; and injection precision. At a minimum, the precision is critical.
Ideally, the standard solutions should be diluted with the dissolution media at a concentration within the linear range of the method, e.g., 100%, or the selected Q value of the dosage strength. However, organic solvent may be used in the preparation of the standard, provided that the accuracy criteria can be met during validation. In some cases, the sample may be diluted with mobile phase to improve the peak shape. The standard and sample solutions should both be prepared in the linear concentration range and measured at the same wavelength.
Automation may require deviations from the pharmacopeial specifications of the instruments, such as incorporation of an integrated outlet on the bottom of the vessel for cleaning and replacement of medium.
Operational steps that are not part of the compendial procedure should be validated. Deviations from the standard procedure described in 711, such as use of sampling probes or fiber-optic probes, should be validated against the standard procedure.
4.1 Medium Preparation
Automated media preparation generally is accomplished by diluting concentrates. Automated media preparation systems typically dispense the volume of medium into the vessel by monitoring either the weight or volume. Chemical and physical stability of the concentrates as well as homogeneity of the dilutions over the intended period of use are important issues and should be understood. Concentrates of buffer solutions and surfactants may have stability issues, such as chemical degradation and pH change. Physical instability may manifest as precipitation, re-crystallization, or phase separation and should be prevented.
If deaeration of the medium is required, the level of deaeration should be specified.
The concentration of the dissolved oxygen can be used to evaluate the efficiency of deaeration procedures discussed in section 2.1 Deaeration.
4.2 Sample Introduction and Timing
Samples should be inserted in the vessel in a reproducible way. Automated sample introduction and aliquot withdrawal provide an advantage over manual sampling because the automated techniques can reduce the variability in the vessel-to-vessel timing of the test intervals. However, automated sample handling may impose timing limitations that need to be considered. The pharmacopeial tolerance of ±2% of the specified dissolution test time may be difficult to meet for early time points.
4.3 Sampling and Filtration
Autosampling is a useful alternative to manual sampling, especially if the test includes several time points. The transfer and filtration of sample solutions from the dissolution instrument to the analytical unit may be undertaken via tube connections or via robotic devices operated in a stepwise procedure. Sample volumes may be removed from the dissolution medium and not returned (consumptive sampling), or the sample volume may be returned to the dissolution medium (recirculated sampling).
There are many brands of autosamplers, including semi-automated and fully automated systems. Routine performance checks, cleaning, and maintenance, as described in the pertinent standard operating procedures or metrology documents, help to ensure reliable operation of these devices.
Sampling probes may or may not remain in the vessel throughout the entire run. Sampling probes or fiber-optic probes can disturb the hydrodynamics of the vessel; therefore, adequate validation should be performed to ensure that the probes are not causing a significant change in the dissolution rate. If filters are used that are different from those used for manual sampling, then these different filters should also be evaluated separately. The position of the pharmacopeial sampling zone for Apparatus 1 and Apparatus 2 is midway from the top of the stirring element to the medium surface and depends on the medium volume. Sampling probes should pull the sample from the sampling zone. Instruments for which the sampling occurs through the hollow shaft should be designed with a means to adjust the depth of the inlet aperture to allow conformance with this requirement. The programmed sampling volume depends on the dead volume of the tubing, cuvettes, and other devices and has to be adjusted accordingly.
A recirculated sampling alignment can be operated either by discharging the tubing contents into the vessel after each sampling or by allowing the tubing to remain filled with solution in the intervals between sampling points. In the latter case, the dead volume and carryover effects are important considerations.
The need for sample volume replacement should be considered. In consumptive sampling with multiple sampling time points, the withdrawn volume may be replaced with an equal volume of fresh medium. The sampling volume may be critical if, in total, it exceeds 1% of the stated volume of dissolution medium required by the procedure. If it can be shown that replacement of the medium is not necessary, the volume change must be part of the calculation of results. See section 2.5 Data Handling.
Carryover may occur when subsequent samples are affected by residues or conditions of previous samples; the effect of the first sample or condition “carries over” to the second. In liquid handling, residues of liquids previously in the sample solution may contaminate subsequent sample solutions. Dissolution media containing surfactants or lipids may present problems. Carryover may occur for successive samples taken over a multiple time-point test, as well as at the beginning of a new test due to the cleaning solution. This topic is discussed in section 4.4 Cleaning.
Interaction of dissolved drug substance with the sampling and transfer devices is an important consideration. When adsorption of the dissolved drug substance occurs, it most often involves surfaces of the dissolution apparatus or sampling filters and tubing. Adsorption may be pH dependent in the case of charged, dissolved drug substance. Adsorption of the dissolved drug to the parts of the sampling device should be assessed using a typical sample solution (dissolution sample from the product or drug substance with formulation matrix) with known concentration. The typical design is a cross-validation with aliquots of the same sample solution passing and bypassing the sampling device (including the sampling probe, filter, tubing, valves, and pump). There is no general recommendation that may give preference to any kind of material or equipment construction (e.g., glass or specific polymers). See section 5.7 Considerations for Automation for more information.
In addition to the information in section 2.4.3 Sampling, connections of pumps and tubing may be sources of contamination in automated systems. Interferences with the spectroscopic analytical procedures, which are commonly used for dissolution testing, are less of a concern. However, interferences must be evaluated if the product under investigation contains low-dose metal salts, as do some dietary supplements.
Liquid transfer usually is undertaken via polymeric tubing. Inert materials such as polytetrafluoroethylene (PTFE) sometimes cannot be used because of their mechanical properties. Where flexible tubes are required, for example in peristaltic pumps or for coiling in a small radius, polypropylene (PP) or high-density polyethylene (HDPE) may be the preferred materials. Depending on the type of polymer and its crystallinity and density, leaching of constituents, mainly plasticizers, may occur. Leachables can interfere with the analytical procedure. The concentration leached to the sample solution usually depends on the surface, the temperature, the exposure time, the hydrodynamic conditions, and the composition of the media.
4.4 Cleaning
In addition to the information in section 2.4.4 Cleaning, automated systems have specific cleaning issues. For example, evaluation of the effectiveness of purging and rinsing between sampling times and within-run condition of the tubing is recommended. Also it is important to evaluate the cleaning process between tests.
4.5 Operating Software and Computation of Results
The software systems for data evaluation and instrument operation must be validated as per 21 CFR 11 (17).
4.6 Common Deviations from the Compendial Procedures That May Require Validation
Some common areas of deviation from compendial procedures include the following:
- Sample introduction relative to start of spindle rotation
- Residence time and positioning of sampling probes
- Recirculated versus consumptive sampling
- Sample volume replacement in consumptive sampling.
The acceptance criteria are presented as guidelines only, and may differ for some products. Manufacturers should document the appropriate acceptance criteria for their products in pertinent Standard Operating Procedures (SOPs) or in validation protocols. Other considerations may be important for special dosage forms. Validation studies should be performed across the range of profile time points. For products containing more than a single active ingredient, the dissolution procedure needs to be validated for each active ingredient. It is expected that investigations into filter suitability and the potential for glass adsorption will have been undertaken already (see 1.1 Performing Filter Compatibility). Validation of these assessments may occur during spiked recovery experiments.
5.1 Specificity/Placebo Interference
It is necessary to demonstrate that the results are not unduly affected by placebo constituents, other active drugs, or degradants. The placebo consists of all the excipients and coatings, with inks and capsule shells included if appropriate, without the active ingredient. Placebo interference can be evaluated by using a spiked placebo that is prepared by weighing samples of the placebo blend, dissolving or dispersing them in dissolution medium at concentrations that would be encountered during testing, and adding a known amount of the drug in solution. It may be preferable to perform this experiment at 37, comparing the solution to a standard solution at the concentration expected to be encountered during testing, by using the formula:
Result = (AP/AS) × CS × (V/L) × 100
AP = absorbance of the placebo
AS = absorbance of the standard
CS = concentration of the standard (mg/mL)
V = volume of the medium (mL)
L = label claim (mg)
The interference should not exceed 2%. Note that for extended-release products, a placebo version of the finished dosage form may be more appropriate than blends because this placebo formulation will release the various excipients in a manner more nearly reflecting the product than will a simple blend of the excipients. In this case, it may be appropriate to evaluate potential interference at multiple sampling points in the release profile, with worst-case interference expected at the later sampling points.
The blank is the dissolution medium without dissolved sample, and it is treated in the same manner as the sample. The effect of the absorbance of the blank at the analytical wavelength should be evaluated. In most cases, the absorbance of the dissolution medium blank may not exceed 1% of the standard solution at the concentration used for analysis. Values >1% should be evaluated on a case-by-case basis.
If the placebo interference exceeds 2%, modification of the method may be necessary. Possible modifications include choosing another wavelength, subtracting baseline using a longer wavelength, transforming absorbance values (e.g., first derivative), and using an alternative analytical technique such as HPLC. Other means for minimizing the placebo interference would be acceptable with appropriate justification. When other active drug substances or significant levels of degradants are present, it is necessary to show that these do not significantly affect the results. One procedure for doing this is to measure the matrix in the presence and absence of the other active drug substance or degradant: any interference should not exceed 2%. Similar approaches may be used if other techniques are used for the analytical finish.
5.2 Linearity and Range
Linearity is typically established by preparing solutions of the drug substance, ranging in concentration from less than the lowest expected concentration to more than the highest concentration during release. The solutions may be prepared either using either a standard solution or spiked solution or by the method of standard addition. A minimum of five concentrations is normally used (see 1225). Typically, solutions are made from a common stock if possible. The concentration range may not exceed the linearity limits of the method, including the instrumentation. Organic solvents may be used to enhance drug solubility for the preparation of the linearity standard solutions. However, no more than 5% (v/v) of organic solvent should be present in the final solution unless validated. Linearity is typically calculated by using an appropriate least-squares regression program. Typically, a square of the correlation coefficient (r2 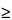 0.98) demonstrates linearity. In addition, the y-intercept must not be importantly different from zero.
0.98) demonstrates linearity. In addition, the y-intercept must not be importantly different from zero.
The range of the procedure is the interval between the upper and lower concentrations of the drug substance (including these levels) that has been demonstrated to have a suitable level of precision, accuracy, and linearity using the procedure as written.
5.3 Accuracy/Recovery
Accuracy/recovery is typically established by preparing multiple samples containing the drug substance and any other constituents present in the dosage form (e.g., excipients, coating materials, capsule shell) ranging in concentration from less than the lowest expected concentration to more than the highest concentration during release. Accuracy/recovery may be done in conjunction with linearity determination. The method of standard addition can also be used. Before this activity, it is expected that filter assessment will already have been performed, and adsorption of drug onto the glass has also been investigated and ruled out.
Individual solutions may be directly prepared in the dissolution medium. Alternatively, to enhance drug solubility it may be appropriate to prepare a stock solution by dissolving the drug substance in a small amount of organic solvent (typically not exceeding 5% organic solvent in the final dissolution media) and diluting to the final concentration with dissolution medium. An amount of stock solution equivalent to the targeted label claim may be used instead of the drug substance powder. Similarly, for very low strengths, it may be more appropriate to prepare a stock solution than to attempt to weigh very small amounts.
The measured recovery is typically 95%–105% of the amount added. Bracketing or matrixing of multiple strengths may be useful. A special case for validation is the Acid Stage procedure described in 711, Delayed-Release Dosage Forms. The limit of NMT 10% needs to be validated. Recovery experiments for drugs that have low solubility in acidic media may be challenging or impossible to perform and may need to be addressed on a case-by-case basis. If the compound degrades in acid, the validation experiment must address this fact.
5.4 Precision
5.4.1 repeatability of analysis
For the analytical finish, repeatability is evaluated by obtaining replicate measurements of standard and/or spiked placebo/standard addition solutions. It can be determined by calculating the RSD of the multiple injections or spectrophotometric readings for each standard solution, or by using the accuracy or linearity data. ICH guidance, Validation of Analytical Procedures: Methodology, recommends that repeatability should be assessed using a minimum of nine determinations covering the specified range for the procedure (i.e., three concentrations and three replicates of each concentration) or using a minimum of six determinations at 100% of the test concentration. A typical acceptance criterion is an RSD of <2%. The demonstration of the repeatability for the dissolution step is conducted by performing the dissolution step on separate units of a well-characterized dosage form or equivalent composite.
5.4.2 intermediate precision/ruggedness
Assuming that the major contributor to the variance is from the dissolution step, intermediate precision may be evaluated to determine the effects of random events on the precision of the dissolution procedure. This evaluation is typically done later in the development of the drug product and is required for full method validation. For many analytical procedures intermediate precision is typically assessed by determination of contributions to variance and, possibly, by a comparison of means. The use of an experimental matrix design is encouraged for evaluation of intermediate precision because interaction effects may be observed more clearly relative to a single variable experiment. In dissolution testing, a ruggedness approach that compares means alone is often taken to investigate the factors that contribute to intermediate precision. The ruggedness can be evaluated across the range of product strengths. Typical variations to be studied include different days, analysts, and equipment. If possible, ruggedness can be evaluated using a drug product lot if well characterized, for example, by having tight content uniformity and uniform performance, but if this type of lot is not available, a premeasured placebo with active ingredients may be used to investigate the intermediate precision. The use of such a spiked placebo would additionally support the assessment of the contribution of the analytical finish to the observed variability of results.
The dissolution procedure on the same lot of well-characterized dosage form may be run by at least two different analysts from the same laboratory, with each analyst preparing the standard solutions and the medium and following the defined extraction/quantification procedure. Typically, the analysts use different dissolution baths, spectrophotometers or HPLC equipment (including columns), and autosamplers, and they perform the test on different days. Full profiles are assessed where relevant to the product. This procedure may not be necessary at each strength; instead, bracketing with high and low strengths may be acceptable.
Acceptance criteria for intermediate precision or for ruggedness are predetermined. A typical acceptance criterion for ruggedness is that the difference in the mean value for dissolution results between any two conditions, using the same strength, does not exceed an absolute 10% at time points with <85% dissolved and does not exceed 5% for time points >85%. Acceptance criteria may be product specific, and other statistical tests and limits may be used.
5.4.3 reproducibility
Reproducibility follows the general concepts of intermediate precision, but is performed by two different analysts at different labs.
5.5 Robustness
Evaluation of robustness, which assesses the effect of making small, deliberate changes to the dissolution conditions, typically is done later in development of the drug product and is a requirement for full method validation. It is performed using a well-characterized lot of drug product, for example having tight content uniformity and uniform performance. The number of replicates (typically 3 or 6) is dependent on the intermediate precision. All profile points should be evaluated.
Selection of parameters to be varied depends on the dissolution procedure and analysis type. The parameters may include medium composition (e.g., buffer or surfactant concentration, pH, deaeration), volume, agitation rate, sampling time, and temperature. Statistical analysis of the data generated will help determine the extent to which the parameters must be controlled in the method. The robustness assessment is well suited to Design of Experiments (DoE) methodologies to efficiently investigate the impact of the individual parameters and/or their interaction.
Robustness of analytical finish is referenced in 1225. HPLC analysis parameters may include mobile phase composition (percentage organic, buffer concentration, pH), flow rate, wavelength, column temperature, and multiple columns (of the same type). For spectrophotometric analysis, the wavelength may be varied.
5.6 Stability of Standard and Sample Solutions
The standard solution is stored under conditions that ensure stability. The stability of the standard solution is analyzed over a specified period of time (for at least the time of the entire dissolution procedure), using a freshly prepared standard solution at each time interval for comparison. The acceptable range for standard solution stability is influenced by the concentration and is typically between 98% and 102% at the expected final concentration.
The sample solution is typically stored at room temperature. The sample is analyzed over a specified period of time, using the original sample solution response for comparison. The typical acceptable range for sample solution stability may be between 98% and 102%, compared with the initial analysis of the sample solutions. If the solution is not stable, aspects to consider include temperature (refrigeration may be needed), light protection, and container material (plastic or glass).
The procedure may state that the standards and samples need to be analyzed within a time period demonstrating acceptable standard and sample solution stability.
5.7 Considerations for Automation
Automated methods offer opportunities for increased precision and reproducibility; however, bias may be introduced. In particular, the sampling probe and the sample lines warrant attention as places where inaccuracies may occur. Deviations from the procedure described in 711, such as resident sampling probes, sampling through the stirring element shaft (hollow-shaft sampling), or fiber-optic probes, should be validated. Other aspects of automation validation may include carryover of residual drug, effect of an in-residence probe, adsorption of drug, and cleaning and/or rinse cycles. Validation is performed using the automated dissolution system including materials. Therefore, any change in materials will require demonstration of suitability based on the validation attributes that are impacted by the change.
Manual and automated procedures should be compared to evaluate the interchangeability of the procedures. This is done by performing two automated runs at each dosage concentration, using all sampling points, compared to manually sampled runs of the same samples. The effect of the in-resident probe cannot be determined by sampling both ways from the same vessel. Results should be consistent with the requirements for intermediate precision if the procedures are to be considered interchangeable. The difference in the mean value for dissolution results between any two conditions using the same strength should not exceed an absolute 10% at time points with <85% dissolved nor exceed 5% for time points >85%. Acceptance criteria may be product specific, and other statistical tests and limits may be used.
Revalidation may be necessary when the automated system is used with different formulations because of the interaction with excipients. Dissolution media containing surfactants or lipids may require additional validation efforts.
6.1 Immediate-Release Dosage Forms
Although release and stability data are collected during dosage form development, it is common to record the entire dissolution profile or the amount of drug dissolved at specified intervals, such as 10, 20, 30, 40, 50, and 60 min or 15, 30, 45, and 60 min. At registration, dissolution for an immediate-release tablet usually becomes a single-point test. The acceptance criterion and test time are established by evaluating the dissolution profile data. The acceptance criterion for a dissolution test is a function of Q, which is expressed as a percentage of label claim of drug dissolved at a specified time. Typical Q values are in the range of 75%–80% dissolved. Q values in excess of 80% are not generally used because allowance needs to be made for assay and content uniformity ranges.
6.2 Delayed-Release Dosage Forms
The discussion about dissolution of delayed-release dosage forms in 711 focuses on enteric-coated dosage forms, which is the most common delayed-release dosage form. A dissolution test for a delayed-release tablet or capsule is a two-part test, and each part has acceptance criteria. First, the dosage forms are exposed to an acid medium, followed by exposure to a buffer medium. To ensure that the enteric coating performs properly, a “NMT” acceptance criterion is indicated in 711 for the acid stage. The medium used for an acid stage is usually 0.1 N HCl, and the duration of this stage is typically 2 h. The dosage forms are then exposed to a buffer medium, usually 0.05 M phosphate buffer at pH 6.8, but other buffers and pH targets may be used if justified. The duration of the buffer stage is usually 45 min for compendial tests, but this duration may vary, depending on the drug product. As with immediate-release dosage forms, a Q value and time point are determined by evaluating the entire dissolution profile.
6.3 Extended-Release Dosage Forms
A dissolution test for an extended-release dosage form is generally similar to that used for an immediate- or delayed-release drug product, except that the duration of the test is longer, and at least three time points are specified for pharmacopeial purposes (20). Additional sampling times may be required for drug approval purposes. An early time point, usually 1–2 h, is chosen to show that dose dumping is not probable. An intermediate time point is chosen to define the in vitro release profile of the dosage form, and a final time point is chosen to show essentially complete release of the drug (20). The time points for the test should be determined by evaluating the dissolution profile across the desired test duration. Often, additional time points are obtained during dosage form development to aid with selecting the appropriate time points for the specification or monograph.
As with an immediate- or delayed-release drug product, the acceptance criteria and time points for an extended-release drug product should discriminate between an acceptable and an unacceptable batch. The acceptance criteria for the first stage of testing (L1) should be established on the basis of available batch data (19,20). If human bioavailability data are available for formulations exhibiting different release rates, then an in vitro/in vivo relationship may be used to establish acceptance criteria (19,20). Acceptance criteria for the second (L2) and third (L3) stages are derived from the L1 criteria using Acceptance Table 2 in 711.
6.4 Multiple Dissolution Tests
Typically, monographs for extended-release dosage forms contain multiple dissolution tests representing specific products. In accordance with General Notices and Requirements 4.10.10, the appropriate test, if not Test 1, is indicated on the product labeling. For example, the USP monograph for Oxycodone Hydrochloride Extended-Release Tablets (21) lists two dissolution tests, each of which has either three or four time points. If the Tablets are analyzed using Test 2 and the dissolution results comply with the criteria provided in the monograph, the labeling for Tablets can indicate that the Tablets meet USP Dissolution Test 2. Multiple dissolution tests also can be found in monographs for immediate- and delayed-release dosage forms. For example, the USP monographs for Levothyroxine Sodium Tablets and Pantoprazole Sodium Delayed-Release Tablets provide four dissolution tests (22,23).
6.5 Interpretation of Dissolution Results
The Interpretation section of 711 discusses immediate-, delayed-, and extended-release dosage forms. The discussion for each of these release patterns is expanded here with examples to assist with applying the criteria during the various stages of testing. Understanding how these criteria are applied will assist in setting appropriate acceptance criteria.
6.5.1 immediate-release dosage forms
Once the Q value is established, the dissolution test is a staged test of three levels. In the first level of testing called S1, six dosage forms are tested. Each dosage form must be Q + 5% (absolute percentage points) dissolved at a specified time. For example, the time and tolerances in a monograph would be:
Time: 30 min
Tolerances: NLT 80% (Q) of the labeled amount of “drug substance” is dissolved.
If the Q value for a 200-mg label claim (LC) immediate-release tablet is specified as 80% and the time point is 30 min, then NLT 85% LC (170 mg) of the drug substance in each tablet must be dissolved at 30 min.
If this criterion is not met, then 6 additional tablets are tested at level 2 (S2). To pass the S2 acceptance criteria, the average of all 12 tablets must be equal to or greater than Q (80% LC; 160 mg in the above example), and no tablet has less than Q – 15% (65% LC; 130 mg in the above example).
If these criteria are not met, then level 3 or S3 testing must be performed by testing 12 additional tablets. To pass S3, the average of all 24 tablets must be equal to or greater than Q (80% LC; 160 mg in the above example). Two additional criteria must be met as well: 1) no more than 2 tablets are less than Q – 15% (65% LC; 130 mg in the above example), and 2) no tablet is less than Q – 25% dissolved (55% LC; 110 mg in the above example.)
6.5.2 delayed-release dosage forms
An aliquot of the acid medium from each vessel is analyzed at the end of the acid stage. For the acid stage, the acceptance criteria have three levels. Level 1 (A1) testing is passed if no individual value exceeds 10% dissolved. If the A1 criteria are not met, then the dissolution test is performed on 6 additional dosage forms for level 2 (A2) testing. Level A2 criteria are passed if the average of all 12 dosage forms in the acid stage is NMT 10% dissolved and if no individual dosage form is more than 25% dissolved. Level 3 testing is performed if the A2 criteria are not met. The A3 criteria are passed if the average of all 24 dosage forms in the acid stage is NMT 10% dissolved and if no individual tablet is more than 25% dissolved. For the special case in which the solubility of the drug in an acidic medium because of conversion to the free acid is too low to support an acceptance criterion of not more than 10% the drug product should be exposed to the acid stage for the defined duration and then exposed to the buffered medium. Alternate acceptance criteria for the acid stage based on drug solubility may be justified.
For delayed-release dosage forms, the total percentage dissolved is determined by adding the measured amounts in the acid and buffer phases for each individual dosage form. These calculated values are then compared to staged acceptance criteria (B1, B2, and B3) that are based on a Q value. The B1, B2, and B3 criteria are identical to those for the immediate release S1, S2, and S3 criteria.
6.5.3 extended-release dosage forms
In the following hypothetical example, which is used to describe the criteria for an extended-release dosage form, the time points are 1, 4, and 8 h. The acceptance range for each time point is as follows:
- Between 24% and 44% LC drug substance dissolved at 1 h
- Between 56% and 76% LC drug substance dissolved at 4 h
- NLT 85% LC drug substance dissolved at 8 h.
Acceptance ranges are often expressed in tabular form in the USP–NF (see Table 3):
Table 3. L1 Criteria
| Time (h) |
Amount Dissolved |
|---|---|
| 1 | 24%–44% |
| 4 | 56%–76% |
| 8 | NLT 85% |
Six tablets are analyzed at Level 1 (L1); acceptance criteria are met if no individual value lies outside each of the stated ranges, and no individual value is less than the percentage specified for the final time point. If the L1 criteria are not met, then 6 additional tablets are analyzed at level 2 (L2). The L2 criteria are met if these three conditions are met:
- The average value of the 12 tablets lies within each of the stated ranges and is NLT the stated range of the final time point.
- None of the 12 tablets is >10% of the labeled content outside each of the stated ranges.
- None of the 12 tablets is >10% of the labeled content below the stated amount at the final test time.
For the above example, the L2 acceptance criteria for the 12 tablets (see Table 4) are as follows:
Table 4. L2 Criteria
| 1 h | 4 h | 8 h | |
|---|---|---|---|
| Average | 24%–44% | 56%–76% | NLT 85% |
| Individual tablets | 14%–54% | 46%–86% | NLT 75% |
If the L2 criteria are not met, then 12 additional tablets are tested at level 3 (L3). The L3 criteria are met if these five conditions are met:
- The average value of the 24 tablets lies within each of the stated ranges and is NLT the stated range of the final time point.
- NMT 2 of the 24 tablets are >10% of labeled content outside each of the stated ranges.
- NMT 2 of the 24 tablets are >10% of the labeled content below the stated amount at the final test time.
- None of the 24 tablets is >20% of the labeled content outside each of the stated ranges.
- None of the 24 tablets is >20% of the labeled content below the stated amount at the final test time.
The L3 acceptance criteria for the 24 tablets in the above example are summarized in Table 5:
Table 5. L3 Criteria
| 1 h | 4 h | 8 h | |
|---|---|---|---|
| Average | 24%–44% | 56%–76% | NLT 85% |
| Individual Tablets | NMT 2 tablets are outside the range of 14%–54%, and no individual tablet is outside the range of 4%–64% | NMT 2 tablets are outside the range of 46%–86%, and no individual tablet is outside the range of 36%–96% | NMT 2 tablets release <75% and no individual tablet releases <65% |
- FDA. Recommended dissolution methods. http://www.accessdata.fda.gov/scripts/cder/dissolution/index.cfm. Accessed 10 Sep 2014.
- Mihali C, Oprea G, Cical E. Determination of critical micelar concentration of anionic surfactants using surfactants—sensible electrodes. Chem Bull “Politehnica” Univ (Timişoara). 2008; 53(67):1–2.
- Stippler ES. Biorelevant dissolution test methods to assess bioequivalence of drug products [PhD dissertation]. Frankfurt: Johann Wolfgang Goethe University; 2004.
- Koga K, Kusawake Y, Ito Y, Sugioka N, Shibata N, Takeda K. Enhancing mechanism of Labrasol on intestinal membrane permeability of the hydrophilic drug gentamicin sulfate. Eur J Pharm Biopharm. 2006; 64(1):82–91.
- Neugebauer JM. Detergents: an overview. Methods Enzymol. 1990; 182:239–253.
- Harris ELV, Angal S, editors. Protein Purification Applications: A Practical Approach. Oxford, England: IRL Press at Oxford University Press; 1990; 71.
- Helenius A, McCaslin DR, Fries E, Tanford C. Properties of detergents. Methods Enzymol. 1979; 56:734–749.
- Kassel D. Properties of cremophore EL micelles probed by fluorescence. Photochem Photobiol. 1992; 56(4):447–451.
- Rowe RC, Sheskey PJ, Quinn ME, editors. Handbook of Pharmaceutical Excipients. 6th ed. London: APhA; 2003.
- Rosenthal KS, Koussale F. Critical micelle concentration determination of non-ionic detergents with coomassie brilliant blue G-250. Anal Chem. 1983; 55:1115–1117.
- Herrman KW. Non-ionic–cationic micellar properties of dimethyldodecylamine oxide. J Phys Chem. 1962; 66:295–300.
- Leeson LJ, Adair D, Clevenger H, Chiang N. The in vitro development of extended-release solid oral dosage forms. J Pharmacokinet Biopharm. 1985; 13(5):493–514.
- Dressman JB, Amidon GL, Reppas C, Shah VP. Dissolution testing as a prognostic tool for oral drug absorption: immediate release dosage forms. Pharm Res. 1998; 15(1):11–22.
- FDA. Guidance for industry. Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. August 2000. http://www.fda.gov/downloads/Drugs/.../Guidances/ucm070246.pdf. Accessed 14 Jun 2013.
- Kakhi M. Classification of the flow regimes in the flow-through cell. Eur J Pharm Sci. 2009; 37(5):531–544.
- FDA. Guidance for industry. Dissolution testing of immediate release solid oral dosage forms. 1997. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/UCM070237.pdf. Accessed 10 Sep 2014.
- FDA. 21 CFR Part 11. April 2012. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfCFR/CFRSearch.cfm?CFRPart=11. Accessed 14 Jun 2013.
- ICH. Q2(R1). Validation of analytical procedures. 1996. http://www.ich.org/products/guidelines/quality/quality-single/article/validation-of-analytical-procedures-text-and-methodology.html. Accessed 17 Jun 2013.
- ICH. Q6A. Specifications: test procedures and acceptance criteria for new drug substances and new drug products: chemical substances. October 1999. http://www.ich.org/products/guidelines/quality/quality-single/article/specifications-test-procedures-and-acceptance-criteria-for-new-drug-substances-and-new-drug-produc.html. Accessed 14 Jun 2013.
- FDA. Guidance for industry. Extended release oral dosage forms: development, evaluation, and application of in vitro/in vivo correlations. September 1997. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070239.pdf. Accessed 14 Jun 2013.
- USP. USP 36–NF 31, Oxycodone Hydrochloride Extended-Release Tablets. Rockville, MD: USP; 2013:4642–4644.
- USP. USP 36–NF 31, Levothyroxine Sodium Tablets. Rockville, MD: USP; 2013:4109–4110.
- USP. USP 36–NF 31, Pantoprazole Sodium Delayed-Release Tablets. Rockville, MD: USP; 2013:4682–4686.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | William E. Brown
Senior Scientific Liaison (301) 816-8380 |
(GCDF2010) General Chapters - Dosage Forms |
USP38–NF33 Page 1090
USP38–NF33 Supplement : No. 1 Page 7103
Pharmacopeial Forum: Volume No. 40(1)