Ranitidine Oral Solution
» Ranitidine Oral Solution is a solution of Ranitidine Hydrochloride in water. It contains the equivalent of not less than 90.0 percent and not more than 110.0 percent of the labeled amount of ranitidine (C13H22N4O3S).
Packaging and storage—
Preserve in tight, light-resistant containers. Store below 25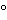 . Do not freeze.
. Do not freeze.
USP Reference standards  11
11 —
—
USP Ranitidine Hydrochloride RS 
') + '&img=../../images/v35300/cas-66357-59-3.gif',600,500);)
USP Ranitidine Related Compound A RS
5-[[(2-Aminoethyl)thio]methyl]-N,N-dimethyl-2-furanmethanamine, hemifumarate salt.
') + '&img=../../images/v35300/c11rs1054.gif',600,500);)
5-[[(2-Aminoethyl)thio]methyl]-N,N-dimethyl-2-furanmethanamine, hemifumarate salt.
USP Ranitidine Related Compound C RS
N-[2-[[[5-[(Dimethylamino)methyl]-2-furanyl]methyl]sulfinyl]ethyl]-N-methyl-2-nitro-1,1-ethenediamine.
') + '&img=../../images/v35300/c11rs1056.gif',600,500);)
N-[2-[[[5-[(Dimethylamino)methyl]-2-furanyl]methyl]sulfinyl]ethyl]-N-methyl-2-nitro-1,1-ethenediamine.
Identification—
A:
The RF value of the principal spot observed in the chromatogram of the Test preparation obtained as directed in the Chromatographic purity test corresponds to that obtained from the Standard preparation.
B:
The retention time of the major peak in the chromatogram of the Assay preparation corresponds to that in the chromatogram of the Standard preparation, as obtained in the Assay.
Microbial enumeration tests 61 and Tests for specified microorganisms 62—
It meets the requirements of the tests for absence of Salmonella species and Escherichia coli; and the total aerobic microbial count does not exceed 100 cfu per mL.
pH 791:
between 6.7 and 7.5.
Chromatographic purity—
Test preparation—
[note—Apply a quantity of extractives from Oral Solution to the chromatographic plate so as to achieve a nominal loading of 200 µg of ranitidine. ] Transfer a weighed quantity of Oral Solution, equivalent to 10 mg of ranitidine, to a suitable syringe. Attach the tip of the syringe to the top of a cartridge (11 mm × 12 mm) of volume 0.5 mL containing 0.4 g of an L1 packing for high-pressure liquid chromatography that has been previously prepared by passage of 10 mL of methanol followed by passage of 20 mL of 0.5 M ammonia solution. Add 2.0 mL of 0.5 M ammonia solution to the syringe and force the mixture slowly through the cartridge. Repeat with 2 further 3-mL portions of 0.5 M ammonia solution. Discard all the liquid that has traversed the cartridge. Pass 5 mL of a mixture of 0.1 M hydrochloric acid and methanol (3:1) through the cartridge, and collect the eluant in a clean round-bottom, 25-mL flask. Repeat this with another 5-mL portion of the same eluting mixture and collect the eluant in the same flask. Evaporate the contents of the flask to dryness at a temperature not exceeding 30. Redissolve the residue in 1.0 mL of a mixture of methanol and water (50:50).
Standard preparation—
Dissolve USP Ranitidine Hydrochloride RS in a mixture of methanol and water (50:50) to obtain a solution having a known concentration of 448 µg (equivalent to 400 µg of ranitidine) per mL. Dilute portions of this Standard preparation quantitatively with the mixture of methanol and water (50:50) to obtain solutions having concentrations of 224 µg per mL (Diluted standard preparation A), 112 µg per mL (Diluted standard preparation B), 56 µg per mL (Diluted standard preparation C), 22 µg per mL (Diluted standard preparation D), and 11 µg per mL (Diluted standard preparation E), respectively.
Resolution preparation—
Dissolve USP Ranitidine Related Compound A RS in methanol to obtain a solution having a known concentration of 1.27 mg per mL.
Procedure—
Apply separately 10 µL of the Standard preparation, the Diluted standard preparations (A, B, C, D, and E), and 20 µL (superposition of 2 × 10 µL) of the Test preparation to a suitable thin-layer chromatographic plate (see Chromatography 621) coated with a 0.25-mm layer of chromatographic silica gel mixture. In addition, apply separately a further loading of 10 µL of the Test preparation to the same plate, and on top of this application, apply 10 µL of the Resolution preparation. Allow the spots to dry, and develop the chromatograms in a solvent system consisting of a mixture of ethyl acetate, isopropyl alcohol, ammonium hydroxide, and water (25:15:5:1) until the solvent front has moved not less than 15 cm from the origin. Remove the plate from the developing chamber, mark the solvent front, and allow to air-dry. Expose the plate to iodine vapors in a closed chamber until the chromatogram is fully revealed. Examine the plate and compare the intensities of any secondary spots observed in the chromatogram of the Test preparation with those of the principal spots in the chromatograms of the Standard preparation and Diluted standard preparations (A, B, C, D, and E): the system suitability requirements are met when there is complete resolution between the primary spots of the Test preparation and the Resolution preparation and if a spot is observed in the chromatogram of Diluted standard preparation E. The major secondary spot is not greater in size or intensity than the principal spot produced by the Standard preparation (2.0%), and no other secondary spot is greater in size or intensity than the principal spot produced by Diluted standard preparation A (1.0%). The sum of the intensities of all secondary spots obtained from the Test preparation corresponds to not more than 5.0%. [note—Spots established as arising from other components in the formulation are to be ignored. ]
Assay—
Mobile phase, Standard preparation, System suitability solution, and Chromatographic system—
Prepare as directed in the Assay under Ranitidine Injection, the chromatographic column being fitted with a suitable pre-column also containing packing L1.
Assay preparation—
Dilute an accurately measured quantity of Oral Solution, quantitatively, and stepwise if necessary, with Mobile phase to obtain a solution having a concentration of 0.1 mg of ranitidine per mL.
Procedure—
Separately inject an equal quantity (about 10 µL) of the Standard preparation and the Assay preparation into the chromatograph, record the chromatograms, and measure the area responses for the major peaks. Calculate the quantity, in mg, of ranitidine (C13H22N4O3S) in the portion of Oral Solution taken by the formula:
(314.40 / 350.87)(L / D)(C)(rU / rS)
in which 314.40 and 350.87 are the molecular weights of ranitidine and ranitidine hydrochloride respectively; L is the labeled quantity of ranitidine in the Oral Solution taken; D is the concentration, in mg per mL, of ranitidine in the Assay preparation, on the basis of the labeled quantity and the extent of dilution; C is the concentration, in mg per mL, of USP Ranitidine Hydrochloride RS in the Standard preparation; and rU and rS are the peak area responses obtained from the Assay preparation and the Standard preparation, respectively.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| Monograph | Elena Gonikberg, Ph.D.
Principal Scientific Liaison 1-301-816-8251 |
(SM32010) Monographs - Small Molecules 3 |
| Reference Standards | RS Technical Services 1-301-816-8129 rstech@usp.org |
|
| Radhakrishna S Tirumalai, Ph.D.
Principal Scientific Liaison 1-301-816-8339 |
(GCM2010) General Chapters - Microbiology | |
| Radhakrishna S Tirumalai, Ph.D.
Principal Scientific Liaison 1-301-816-8339 |
(GCM2010) General Chapters - Microbiology |
USP35–NF30 Page 4523
Pharmacopeial Forum: Volume No. 30(6) Page 2036