Hydroxypropyl Cellulose
Cellulose, 2-hydroxypropyl ether [9004-64-2].
[9004-64-2].
(hye drox'' ee proe' pil sel' ue lose).
Cellulose, 2-hydroxypropyl ether
DEFINITION
Hydroxypropyl Cellulose is a partially substituted poly(hydroxypropyl) ether of cellulose. It may contain NMT 0.60% of silica or other suitable anticaking agents. When dried at 105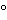 for 1 h, it contains NMT 80.5% of hydroxypropoxy groups.
for 1 h, it contains NMT 80.5% of hydroxypropoxy groups.
IDENTIFICATION
• Infrared Absorption  197K
197K :
[Note—The spectrum may or may not contain a peak at about 1719 cm
:
[Note—The spectrum may or may not contain a peak at about 1719 cm 1. ]
1. ]
ASSAY
• Hydroxypropoxy Groups
Apparatus:
The apparatus for hydroxypropoxy group determinations is shown in Figure 1.
+'&img=../../images/v35300/g-1701.gif&casNumber=9004-64-2&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 1. Apparatus for hydroxypropoxy determination.
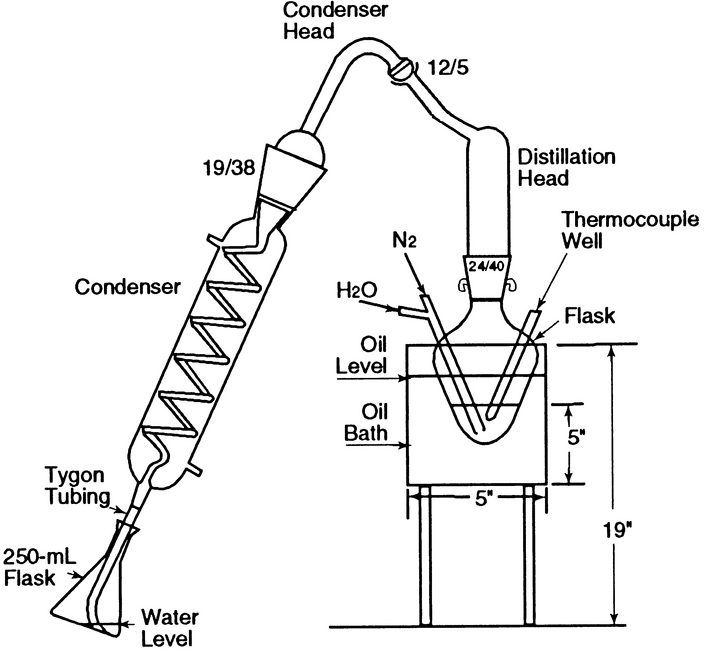
The boiling or reaction flask, consisting of a 125-mL conical-bottom boiling flask modified to provide a thermocouple (or thermometer) well and an inlet with a 1.0-mm capillary tip for nitrogen and water (see Figure 2), is fitted with a distillation head that leads to a condenser. The reaction flask is immersed in an oil bath equipped with an electric heater capable of heating the bath at the desired rate and maintaining the temperature at 155. The distillate is collected in a flask. [Note—The tube from the condenser to the flask must be below the surface of the liquid in the flask to ensure the capture of all of the acetic acid formed. See Figure 1. ]
+'&img=../../images/v35300/g-1702.gif&casNumber=9004-64-2&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 2. Boiling flask.
Analysis:
Transfer 65 mg of Hydroxypropyl Cellulose, previously dried at 105 for 1 h, into the reaction flask. Add 5 mL of water, and swirl gently for 5 min. Add 10 mL of chromium trioxide solution (30 g in 70 mL). Assemble the apparatus as shown in Figures 1 and 2, and immerse the reaction flask in the oil bath slightly above the level of the chromium trioxide solution. Start the condenser cooling water, and pass nitrogen gas through the flask at a rate of about 70–75 mL/min. Raise the temperature of the oil bath to 155 during a 30-min period, and maintain it at this temperature throughout the determination. [Note—Too rapid an initial rise in temperature results in high blank determinations. ]
Monitor the temperature of the reaction mixture in the reaction flask using a thermocouple or thermometer in a well, as shown in Figures 1 and 2. When a reaction mixture temperature of 102 ± 1 is reached, add water through the water inlet until the reaction mixture temperature drops to 97 ± 1. Continue this 97 to 102 temperature cycle until 100 mL of distillate has been collected. Detach the condenser from the distillation head, and wash with water, collecting the washings in the flask containing the distillate. Titrate the solution with 0.02 N sodium hydroxide VS to a pH of 7.0 ± 0.1, using an expanded-scale pH meter equipped with glass and calomel electrodes. Record the volume, V, of the 0.02 N sodium hydroxide used, then add 500 mg of sodium bicarbonate and 10 mL of 2 N sulfuric acid. After evolution of carbon dioxide has ceased, add 1 g of potassium iodide, insert the stopper in the flask, shake the mixture, and allow the solution to stand in the dark for 5 min. Titrate the liberated iodine with 0.02 N sodium thiosulfate VS to the sharp disappearance of the yellow iodine color, adding a few drops of starch TS to confirm the endpoint. Record the volume, Y, required. This titration, Y mL, multiplied by the empirical factor, K, appropriate to the particular apparatus and reagents in use (see the calculation below), gives the acid equivalent not caused by acetic acid. The acetic acid equivalent is (V KY) mL of 0.02 N sodium hydroxide.
Obtain the empirical factor, K, for the apparatus by performing a blank determination in which the Hydroxypropyl Cellulose is omitted. The acidity of the blank for a given apparatus and given reagents is in a fixed ratio to the oxidizing equivalent of the distillate in terms of sodium thiosulfate:
Result = (VB × N1)/(YB × N2)
| VB | = | = volume of 0.02 N sodium hydroxide required in the blank run (mL) |
| N1 | = | = normality of the 0.02 N sodium hydroxide |
| YB | = | = volume of 0.02 N sodium thiosulfate required in the blank run (mL) |
| N2 | = | = normality of the 0.02 N sodium thiosulfate |
Calculate the percentage of hydroxypropoxy groups (–OCH2CHOHCH3):
Result = (VAN1 KYAN2) × (0.079/W) × 100
| VA | = | = volume of 0.02 N sodium hydroxide required for titration of the sample (mL) |
| N1 | = | = normality of the 0.02 N sodium hydroxide |
| K | = | = empirical factor |
| YA | = | = volume of 0.02 N sodium thiosulfate required for titration of the sample (mL) |
| N2 | = | = normality of the 0.02 N sodium thiosulfate |
| W | = | = quantity of sample used (g) |
Each mL of 0.02 N sodium hydroxide is equivalent to 1.502 mg of hydroxypropoxy groups (–OCH2CHOHCH3).
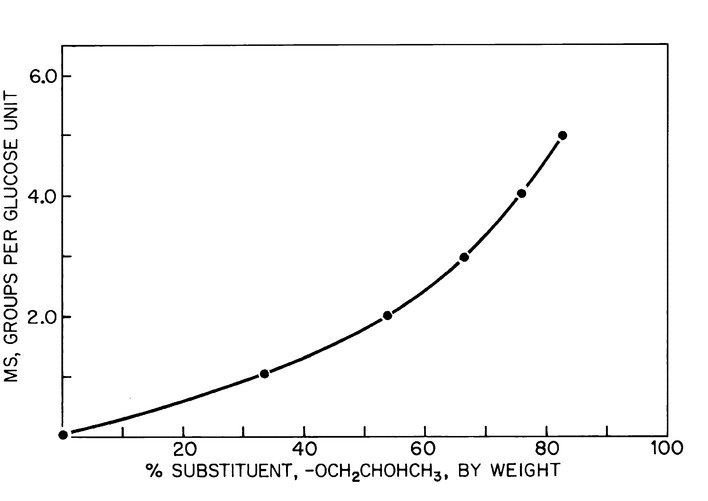
The results obtained as a percentage of hydroxypropoxy content may be converted to terms of average molecular substitution of glucose units by means of the graph in Figure 3.
+'&img=../../images/v35300/g-1703.gif&casNumber=9004-64-2&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 3. Graph for converting percentage of substitution, by weight, of hydroxypropoxy groups to molecular substitution/glucose unit.
Acceptance criteria:
NMT 80.5% of hydroxypropoxy groups
IMPURITIES
Inorganic Impurities
• Residue on Ignition 281
[Caution—Perform the mixing and heating of the mixtures containing hydrofluoric acid in a well-ventilated hood.
]
Analysis:
Proceed as directed for Residue on Ignition 281, using a platinum crucible if silica may be present. If more than 0.2% residue is found, and silica is present, moisten the residue with water, and add 5 mL of hydrofluoric acid, in small portions. Evaporate on a steam bath to dryness, and cool. Add 5 mL of hydrofluoric acid and 0.5 mL of sulfuric acid, and evaporate to dryness. Slowly increase the temperature until all of the acids have been volatilized, and ignite at 1000 ± 25. Cool in a desiccator, and weigh: the difference between the final weight and the weight of the initially ignited portion represents the weight of silica.
Acceptance criteria:
The final weight is NMT 0.2% of the weight of the sample taken for the ignition.
• Lead 251:
NMT 10 ppm
• Heavy Metals, Method II 231:
NMT 20 ppm
SPECIFIC TESTS
• pH 791:
5.0–8.0, in a solution (1 in 100)
• Loss on Drying 731:
Dry a sample at 105 for 3 h: it loses NMT 5.0% of its weight.
• Viscosity 911:
Determine the apparent viscosity at the concentration and temperature specified on the label with a suitable rotational viscosimeter (see Labeling).
ADDITIONAL REQUIREMENTS
• Packaging and Storage:
Store in well-closed containers.
• Labeling:
Label it to indicate the viscosity in an aqueous solution of stated concentration and temperature. The indicated viscosity may be in the form of a range encompassing 50%–150% of the average value.
• USP Reference Standards 11
USP Hydroxypropyl Cellulose RS 
') + '&img=../../images/v35300/cas-9004-64-2.gif',600,500);)
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| Monograph | Kevin T. Moore, Ph.D.
Senior Scientific Liaison 1-301-816-8369 |
(EXC2010) Monographs - Excipients |
| Reference Standards | RS Technical Services 1-301-816-8129 rstech@usp.org |
USP35–NF30 Page 1821
Pharmacopeial Forum: Volume No. 35(3) Page 672