Change to read:
Introduction and General Information
The activity (potency) of antibiotics can be demonstrated by their inhibitory effect on microorganisms under suitable conditions. A reduction in antimicrobial activity may not be adequately demonstrated by chemical methods. This chapter summarizes procedures for the antibiotics recognized in the United States Pharmacopeia (USP) for which the microbiological assay is the standard analytical method.
Two general techniques are employed: the cylinder-plate (or plate) assay and the turbidimetric (or tube) assay. Table 1 lists all the antibiotics that contain microbial assays and specifies the type of assay (cylinder-plate or turbidimetric).
Table 1
| Antibiotic | Type of Assay |
|---|---|
| Amphotericin B | Cylinder-plate |
| Bacitracin | Cylinder-plate |
| Bleomycin | Cylinder-plate |
| Capreomycin | Turbidimetric |
| Carbenicillin | Cylinder-plate |
| Chloramphenicol | Turbidimetric |
| Chlortetracycline | Turbidimetric |
| Cloxacillin | Cylinder-plate |
| Colistemethate | Cylinder-plate |
| Colistin | Cylinder-plate |
| Dihydrostreptomycin | Cylinder-plate |
| Turbidimetric | |
| Erythromycin | Cylinder-plate |
| Gentamicin | Cylinder-plate |
| Gramicidin | Turbidimetric |
| Nafcillin | Cylinder-plate |
| Natamycin | Cylinder-plate |
| Neomycin | Cylinder-plate |
| Turbidimetric | |
| Novobiocin | Cylinder-plate |
| Nystatin | Cylinder-plate |
| Oxytetracycline | Turbidimetric |
| Paromomycin | Cylinder-plate |
| Penicillin G | Cylinder-plate |
| Polymyxin B | Cylinder-plate |
| Sisomicin | Cylinder-plate |
| Tetracycline | Turbidimetric |
| Thiostrepton | Turbidimetric |
| Troleandomycin | Turbidimetric |
| Tylosin | Turbidimetric |
| Vancomycin | Cylinder-plate |
[Note—Perform all procedures under conditions designed to avoid extrinsic microbial contamination. Take adequate safety precautions while performing these assays because of possible allergies to drugs and because live cultures of organisms are used in the procedures. ]
Cylinder-plate assay:
The cylinder-plate assay depends on diffusion of the antibiotic from a vertical cylinder through a solidified agar layer in a Petri dish or plate. The growth of the specific microorganisms inoculated into the agar is prevented in a circular area or zone around the cylinder containing the solution of the antibiotic.
Turbidimetric assay:
The turbidimetric assay depends on the inhibition of growth of a microorganism in a uniform solution of the antibiotic in a fluid medium that is favorable to the growth of the microorganism in the absence of the antibiotic.
Units and Reference Standards:
The potency of antibiotics is designated in either units (U) or µg of activity. In each case the unit or µg of antibiotic activity was originally established against a United States Federal Master Standard for that antibiotic. The corresponding USP Reference Standard is calibrated in terms of the master standard.
Originally, an antibiotic selected as a reference standard was thought to consist entirely of a single chemical entity and was therefore assigned a potency of 1000 µg/mg. In several such instances, as the manufacturing and purification methods for particular antibiotics became more advanced, antibiotics containing more than 1000 µg of activity/mg became possible. Such antibiotics had an activity equivalent to a given number of µg of the original reference standard. In most instances, however, the µg of activity is exactly equivalent numerically to the µg (weight) of the pure substance. In some cases, such as those listed below, the µg of activity defined in terms of the original master standard is equal to a unit:
- Where an antibiotic exists as the free base and in salt form and the µg of activity has been defined in terms of one of these forms
- Where the antibiotic substance consists of a number of components that are chemically similar but differ in antibiotic activity
- Where the potencies of a family of antibiotics are expressed in terms of a reference standard consisting of a single member which, however, might itself be heterogeneous
Do not assume that the µg of activity corresponds to the µg (weight) of the antibiotic substance.
Apparatus:
Labware used for the storage and transfer of test dilutions and microorganisms must be sterile and free of interfering residues (see Cleaning Glass Apparatus  1051
1051 ). Use a validated sterilization method, such as dry heat, steam, or radiation; or use sterile, disposable labware.
). Use a validated sterilization method, such as dry heat, steam, or radiation; or use sterile, disposable labware.
Temperature control:
Thermostatic control is required in several stages of a microbial assay: when culturing a microorganism and preparing its inoculum, and during incubation in plate and tube assays. Refer to specific temperature requirements below for each type of assay.
Test organisms:
The test organism for each antibiotic is listed in Table 3 for the cylinder-plate assay and Table 8 for the turbidimetric assay. The test organisms are specified by the American Type Culture Collection (ATCC) number.
In order to ensure acceptable performance of test organisms, store and maintain them properly. Establish the specific storage conditions during method validation or verification. Discard cultures if a change in the organism's characteristics is observed.
Prolonged storage:
For prolonged storage, maintain test organisms in a suitable storage solution such as 50% fetal calf serum in broth, 10%–15% glycerol in tryptic soy broth, defribinated sheep blood, or skim milk. Prolonged-storage cultures are best stored in the freeze-dried state; temperatures of 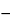 60
60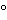 or below are preferred; temperatures below 20 are acceptable.
or below are preferred; temperatures below 20 are acceptable.
Primary cultures:
Prepare primary cultures by transferring test organisms from prolonged-storage vials onto appropriate media, and incubate under appropriate growth conditions. Store primary cultures at the appropriate temperature, usually 2–8, and discard after three weeks. A single primary culture can be used to prepare working cultures only for as many as seven days.
Working cultures:
Prepare working cultures by transferring the primary culture onto appropriate solid media to obtain isolated colonies. Incubate working cultures under appropriate conditions to obtain satisfactory growth for preparation of test inocula. Prepare fresh working cultures for each test day.
Uncharacteristic growth or performance of a test organism:
Use new stock cultures, primary cultures, or working cultures when a test organism shows uncharacteristic growth or performance.
Assay designs:
Suitable experimental designs are key to increasing precision and minimizing bias. Control of the incubation parameters, temperature distribution and time, is critical for minimizing bias; it can be accomplished by staging the plates and racks as described for each assay.
Cylinder-plate assay:
The comparisons are restricted to relationships between zone diameter measurements within plates, excluding the variation between plates. Individual plate responses are normalized on the basis of the relative zone size of the standard compared to the mean zone size of the standard across all plates.
Turbidimetric assay:
To avoid systematic bias, place replicate tubes randomly in separate racks so that each rack contains one complete set of treatments. The purpose of this configuration is to minimize the influence of temperature distribution on the replicate samples. The turbidimetric assay, because of the configuration of the samples in test tube racks, is sensitive to slight variations in temperature. The influence of temperature variation can also be decreased by ensuring proper airflow or heat convection during incubation. At least three tubes for each sample and standard concentration (one complete set of samples) should be placed in a single rack. The comparisons are restricted to relationships between the observed turbidities within racks.
Potency considerations:
Within the restrictions listed above, the recommended assay design employs a five-concentration standard curve and a single concentration of each sample preparation.
For the cylinder-plate assay, each plate includes only two treatments, the reference treatment (median level standard, i.e., S3) and one of the other four concentrations of the standard (S1, S2, S4, and S5) or the sample (U3). The concentration of the sample is an estimate based on the target concentration. The sample should be diluted to give a nominal concentration that is estimated to be equivalent to the median reference concentration (S3) of the standard. The purpose of diluting to the median reference concentration is to ensure that the sample result will fall within the linear portion of the standard curve. The test determines the relative potency of U3 against the standard curve. The sample (U3) should have a relative potency of about 100%. The final potency of the sample is obtained by multiplying the U3 result by the dilution factor.
An assay should be considered preliminary if the computed potency value of the sample is less than 80% or more than 125%. In this case, the results suggest that the sample concentration assumed during preparation of the sample stock solution was not correct. In such a case, one can adjust the assumed potency of the sample on the basis of the preliminary potency value and repeat the assay. Otherwise, the potency will be derived from a portion of the curve where the standard and sample responses will likely not be parallel.
Microbial determinations of potency are subject to inter-assay as well as intra-assay variables; therefore two or more independent assays are required for a reliable estimate of the potency of a given sample. Starting with separately prepared stock solutions and test dilutions of both the standard and the sample, perform additional assays of a given sample on a different day. The mean potency should include the results from all the valid independent assays. The number of assays required in order to achieve a reliable estimate of potency depends on the variability of the assay and the required maximum uncertainty for the potency estimate. The latter is assessed by the width of the confidence interval (refer to Calculations, Confidence limits and combinations of assay calculations). The combined result of a series of smaller, independent assays spread over a number of days is a more reliable estimate of potency than one from a single large assay with the same total number of plates or tubes. Note that additional assays or lower variability allows the product to meet tighter specification ranges. Reducing assay variability achieves the required confidence limit with fewer assays.
Cylinder-Plate Method
Temperature control:
Use appropriately qualified and calibrated equipment to obtain the temperature ranges specified in Table 3.
Apparatus
Plates:
Glass or disposable plastic Petri dishes (approximately 20 × 100 mm or other appropriate dimensions) with lids
Cylinders:
Stainless steel or porcelain cylinders; 8 ± 0.1-mm o.d.; 6 ± 0.1-mm i.d.; 10 ± 0.1-mm high. [Note—Carefully clean cylinders to remove all residues; occasional cleaning in an acid bath, e.g., with about 2 N nitric acid or with chromic acid (see Cleaning Glass Apparatus 1051) is required. ]
Standard solutions:
To prepare a stock solution, dissolve a suitable quantity of the USP Reference Standard of a given antibiotic, or the entire contents of a vial of USP Reference Standard, where appropriate, in the solvent specified in Table 2; and dilute to the specified concentration. Store at 2–8, and use within the period indicated. On the day of the assay, prepare from the stock solution five or more test dilutions, in which the successive solutions increase stepwise in concentration, usually in the ratio of 1:1.25. Use the final diluent specified such that the median has the concentration suggested in Table 2.
Sample solutions:
Assign an assumed potency per unit weight or volume to the sample. On the day of the assay prepare a stock solution in the same manner specified for the USP Reference Standard (Table 2). Dilute the sample stock solution in the specified final diluent to obtain a nominal concentration equal to the median concentration of the standard (S3).
Table 2
| Antibiotic | Stock Solution | Test Dilution | |||||
|---|---|---|---|---|---|---|---|
| Initial Solvent |
Initial Concentration |
Further Diluent |
Final Concentration |
Use Within |
Final Diluent |
Median Concentration (S3)a,b |
|
| Amphotericin Bc,d | Dimethyl sulfoxide | — | — | 1 mg/mL | Same day | B.10e | 1 µg/mL |
| Bacitracinf | 0.01 N hydrochloric acid | — | — | 100 U/mL | Same day | B.1e | 1 U/mL |
| Bleomycin | B.16e | — | — | 2 U/mL | 14 days | B.16e | 0.04 U/mL |
| Carbenicillin | B.1e | — | — | 1 mg/mL | 14 days | B.1e | 20 µg/mL |
| Cloxacillin | B.1e | — | — | 1 mg/mL | 7 days | B.1e | 5 µg/mL |
| Colistemethatec | Water | 10 mg/mL | B.6e | 1 mg/mL | Same day | B.6e | 1 µg/mL |
| Colistin | Water | 10 mg/mL | B.6e | 1 mg/mL | 14 days | B.6e | 1 µg/mL |
| Dihydrostreptomycing | B.3e | — | — | 1 mg/mL | 30 days | B.3e | 1 µg/mL |
| Erythromycin | Methanol | 10 mg/mL | B.3e | 1 mg/mL | 14 days | B.3e | 1 µg/mL |
| Gentamicin | B.3e | — | — | 1 mg/mL | 30 days | B.3e | 0.1 µg/mL |
| Nafcillin | B.1e | — | — | 1 mg/mL | 2 days | B.1e | 2 µg/mL |
| Natamycin | Dimethyl sulfoxide | — | — | 1 mg/mL | Same day | B.10e | 5 µg/mL |
| Neomycing | B.3e | — | — | 1 mg/mL | 14 days | B.3e | 1 µg/mL |
| Novobiocin | alcohol | 10 mg/mL | B.3e | 1 mg/mL | 5 days | B.6e | 0.5 µg/mL |
| Nystatinc, h | Dimethylformamide | — | — | 1000 U/mL | Same day | B.6e | 20 U/mL |
| Paromomycin | B.3e | — | — | 1 mg/mL | 21 days | B.3e | 1 µg/mL |
| Penicillin G | B.1e | — | — | 1000 U/mL | 4 days | B.1e | 1 U/mL |
| Polymyxin Bi | Water | — | B.6e | 10,000 U/mL |
14 days | B.6e | 10 U/mL |
| Sisomicin | B.3e | — | — | 1 mg/mL | 14 days | B.3e | 0.1 µg/mL |
| Vancomycin | Water | — | — | 1 mg/mL | 7 days | B.4e | 10 µg/mL |
|
a
It is acceptable to adjust the median concentration to optimize zone sizes if the data remain in the linear range.
b
µg in this column refers to µg of activity.
c
Prepare the USP Reference Standard and sample test dilutions simultaneously.
d
Further dilute the stock solution with dimethyl sulfoxide to give concentrations of 12.8, 16, 20, 25, and 31.2 µg/mL before making the test dilutions.The test dilution of the sample should contain the same amount of dimethyl sulfoxide as the test dilutions of the USP Reference Standard.
e
The letter B refers to buffer. See Media and Solutions, Buffers for a description of each buffer listed in this table.
f
Each of the standard test dilutions should contain the same amount of hydrochloric acid as the test dilution of the sample.
g
The turbidimetric assay can be used as an alternative procedure.
h
Further dilute the stock solution with dimethylformamide to give concentrations of 256, 320, 400, 500, and 624 U/mL before making the test dilutions. Prepare the standard test dilutions simultaneously with test dilutions of the sample to be tested. The test dilution of the sample should contain the same amount of dimethylformamide as the test dilutions of the standard. Use low-actinic glassware.
i
Prepare the stock solution by adding 2 mL of water for each 5 mg of the USP Reference Standard.
|
|||||||
Inocula:
Suspend the test organism from a freshly grown slant or culture in 3 mL of sterile saline TS. Glass beads can be used to facilitate the suspension. Spread the saline suspension onto the surface of two or more agar plates (covering the entire surface) or onto the surface of a Roux bottle containing 250 mL of the specified medium (see Table 3).
Incubate for the specified time and at the temperature as specified in Table 3, or until growth is apparent.
After incubation, harvest the organism from the plates or Roux bottle with approximately 50 mL of sterile saline TS (except use Medium 34 for bleomycin; see the section Media and Solutions), using a sterile bent glass rod or sterile glass beads. Pipet the suspension into a sterile glass container. This is the harvest suspension.
Dilute an appropriate amount of the harvest suspension with sterile saline TS. Using the UV-visible spectrophotometer, measure % transmittance at 580 nm. The target value is approximately 25% transmittance at 580 nm. This value is used to standardize the harvest suspension volume added to the seed layer agar.
Starting with the suggested volumes indicated in Table 3, determine during method verification the proportions of stock suspension to be added to the inoculum medium that result in satisfactory zones of inhibition of approximately 14–16 mm in diameter for the median concentration of the standard (S3). [Note—Zone sizes that are outside the 11 to 19-mm range are not desirable, because these contribute to assay variability. ] If the dilution percentage transmittance is above 25%, a ratio may be used to normalize the addition of organism to the seed layer. The normalization factor can be determined by dividing the percentage transmittance obtained from the dilution by 25. This ratio can then be multiplied by the suggested inoculum amount to obtain the volume (mL) of harvest suspension that needs to be added to the seed layer. Adjust the quantity of inoculum on a daily basis, if necessary, to obtain an optimum concentration–response relationship.
Alternatively, determine during method verification the proportion of harvest suspension to be incorporated into the inoculum, starting with the volumes indicated in Table 3, that result in satisfactory demarcation of the zones of inhibition of about 14–16 mm in diameter for the median concentration of the standard (S3) and giving a reproducible concentration–response relationship. Prepare the inoculum by adding a portion of stock suspension to a sufficient amount of agar medium that has been melted and cooled to 45–50. Swirl the mixture without creating bubbles in order to obtain a homogeneous suspension.
Table 3
| Antibiotic | Test Organism | ATCCa
Number |
Incubation Conditions | Suggested Inoculum Composition |
|||
|---|---|---|---|---|---|---|---|
| Mediumb | Temperature ( |
Time | Mediumb | Amount (mL/100 mL) |
|||
| Amphotericin B | Saccharomyces cerevisiae | 9763 | 19 | 29–31 | 48 h | 19 | 1.0 |
| Bacitracin | Micrococcus luteus | 10240 | 1 | 32–35 | 24 h | 1 | 0.3 |
| Bleomycin | Mycobacterium smegmatis | 607 | 36 | 36–37.5 | 48 h | 35 | 1.0 |
| Carbenicillinc | Pseudomonas aeruginosa | 25619 | 1 | 36–37.5 | 24 h | 10 | 0.5 |
| Cloxacillin | Staphylococcus aureus | 29737 | 1 | 32–35 | 24 h | 1 | 0.1 |
| Colistimethate | Bordetella bronchiseptica | 4617 | 1 | 32–35 | 24 h | 10 | 0.1 |
| Colistin | Bordetella bronchiseptica | 4617 | 1 | 32–35 | 24 h | 10 | 0.1 |
| Dihydrostreptomycin | Bacillus subtilis | 6633 | 32 | 32–35 | 5 days | 5 | As required |
| Erythromycin | Micrococcus luteus | 9341 | 1 | 32–35 | 24 h | 11 | 1.5 |
| Gentamicin | Staphylococcus epidermidis | 12228 | 1 | 32–35 | 24 h | 11 | 0.03 |
| Nafcillin | Staphylococcus aureus | 29737 | 1 | 32–35 | 24 h | 1 | 0.3 |
| Neomycin | Staphylococcus epidermidis | 12228 | 1 | 32–35 | 24 h | 11 | 0.4 |
| Novobiocin | Staphylococcus epidermidis | 12228 | 1 | 32–35 | 24 h | 1 | 4.0 |
| Nystatin | Saccharomyces cerevisiae | 2601 | 19 | 29–31 | 48 h | 19 | 1.0 |
| Paromomycin | Staphylococcus epidermidis | 12228 | 1 | 32–35 | 24 h | 11 | 2.0 |
| Penicillin G | Staphylococcus aureus | 29737 | 1 | 32–35 | 24 h | 1 | 1.0 |
| Polymyxin B | Bordetella bronchiseptica | 4617 | 1 | 32–35 | 24 h | 10 | 0.1 |
| Sisomicin | Staphylococcus epidermidis | 12228 | 1 | 32–35 | 24 h | 11 | 0.03 |
| Vancomycin | Bacillus subtilis | 6633 | 32 | 32–35 | 5 days | 8 | As required |
|
a
American Type Culture Collection, 10801 University Boulevard, Manassas VA 20110-2209 (http://www.atcc.org)
b
See Media and Solutions, Media.
c
Use 0.5 mL of a 1:25 dilution of the stock suspension/100 mL of Medium 10.
|
|||||||
Analysis:
Prepare the base layer for the required number of assay Petri plates, using the medium and volume shown in Table 4. Allow it to harden into a smooth base layer of uniform depth. Prepare the appropriate amount of seed layer inoculum (Table 5) as directed for the given antibiotic (Table 3) with any adjustments made based on the preparatory trial analysis. Tilt the plate back and forth to spread the inoculum evenly over the base layer surface, and allow it to harden.
Table 4 (base layer)
| Antibiotic | Mediuma | Target Volume (mL) |
|---|---|---|
| Amphotericin Bb | — | — |
| Bleomycin | 35 | 10 |
| Carbenicillin | 9 | 21 |
| Colistimethate | 9 | 21 |
| Colistin | 9 | 21 |
| Dihydrostreptomycin | 5 | 21 |
| Erythromycin | 11 | 21 |
| Gentamicin | 11 | 21 |
| Neomycin | 11 | 21 |
| Nystatinb | — | — |
| Paromomycin | 11 | 21 |
| Polymyxin B | 9 | 21 |
| Sisomicin | 11 | 21 |
| Vancomycin | 8 | 10 |
| All others | 2 | 21 |
|
a
See Media and Solutions, Media.
b
No base layer is used.
[Note—The base layer may be warmed to facilitate a uniform seed layer. ] |
||
Table 5 (seed layer)
| Antibiotic | Mediuma | Target Volume (mL) |
|---|---|---|
| Amphotericin B | Refer to Table 3 | 8 |
| Bleomycin | 6 | |
| Nystatin | 8 | |
| All others | 4 | |
|
a
See Media and Solutions, Media.
|
||
Drop six assay cylinders on the inoculated surface from a height of 12 mm, using a mechanical guide or other device to ensure even spacing on a radius of 2.8 cm, and cover the plates to avoid contamination. Fill the six cylinders on each plate with dilutions of antibiotic containing the test levels (S1–S5 and U3) specified in the following paragraph. Incubate the plates as specified in Table 6 for 16–18 h, and remove the cylinders. Measure and record the diameter of each zone of growth inhibition to the nearest 0.1 mm.
Table 6
| Antibiotic | Incubation Temperature ( |
|---|---|
| Amphotericin B | 29–31 |
| Carbenicillin | 36–37.5 |
| Colistimethate | 36–37.5 |
| Colistin | 36–37.5 |
| Dihydrostreptomycin | 36–37.5 |
| Gentamicin | 36–37.5 |
| Neomycin | 36–37.5 |
| Novobiocin | 34–36 |
| Nystatin | 29–31 |
| Paromomycin | 36–37.5 |
| Polymyxin B | 36–37.5 |
| Sisomicin | 36–37.5 |
| Vancomycin | 36–37.5 |
| All others | 32–35 |
The standards (S1–S5) and a single test level of the sample (U3) corresponding to S3 of the standard curve, as defined in Standard solutions and Sample solutions will be used in the assay. For deriving the standard curve, fill alternate cylinders on each of three plates with the median test dilution (S3) of the standard and each of the remaining nine cylinders with one of the other four test dilutions of the standard. Repeat the process for the three test dilutions of the standard. For the sample, fill alternate cylinders on each of three plates with the median test dilution of the standard (S3), and fill the remaining nine cylinders with the corresponding test dilution (U3) of the sample.
Turbidimetric Method
Temperature control:
Use appropriately qualified and calibrated equipment to obtain the temperature ranges specified in Table 8. [Note—Temperature control can be achieved using either circulating air or water. The greater heat capacity of water lends it some advantage over circulating air. ]
Spectrophotometer:
Measuring absorbance or transmittance within a fairly narrow frequency band requires a suitable spectrophotometer in which the wavelength can be varied or restricted by use of 580-nm or 530-nm filters. Alternatively, a variable-wavelength spectrophotometer can be used and set to a wavelength of 580 nm or 530 nm.
The instrument may be modified as follows:
- To accept the tube in which incubation takes place (see Apparatus below)
- To accept a modified cell fitted with a drain that facilitates rapid change of contents
- To contain a flow cell for a continuous flowthrough analysis
Autozero the instrument with clear, uninoculated broth prepared as specified for the particular antibiotic, including the same amount of test dilution (including formaldehyde if specified) as found in each sample.
Either absorbance or transmittance can be measured while preparing inocula.
Apparatus:
Glass or plastic test tubes, e.g., 16 × 125 mm or 18 × 150 mm. [Note—Use tubes that are relatively uniform in length, diameter, and thickness and substantially free from surface blemishes and scratches. In the spectrophotometer, use matched tubes that are free from scratches or blemishes. Clean tubes thoroughly to remove all antibiotic residues and traces of cleaning solution. Sterilize tubes before use. ]
Standard solutions:
To prepare a stock solution, dissolve a quantity of the USP Reference Standard of a given antibiotic or the entire contents of a vial of USP Reference Standard, where appropriate, in the solvent specified in Table 7, and dilute to the required concentration. Store at 2–8, and use within the period indicated. On the day of the assay, prepare from the stock solution five or more test dilutions, the successive solutions increasing stepwise in concentration, usually in the ratio of 1:1.25. [Note—It may be necessary to use smaller ratios for the successive dilutions from the stock solution for the turbidimetric assay. ] Use the final diluent specified such that the median level of the standard (S3) has the concentration suggested in Table 7.
Sample solutions:
Assign an assumed potency per unit weight or volume to the unknown, and on the day of the assay prepare a stock solution in the same manner specified for the USP Reference Standard (Table 7). Dilute the sample stock solution in the specified final diluent at a nominal concentration equal to the median concentration of the standard (S3) as specified in Table 7.
Table 7
| Antibiotic | Stock Solution | Test Dilution | |||||
|---|---|---|---|---|---|---|---|
| Initial Solvent |
Initial Concentration |
Further Diluent |
Final Stock Concentration |
Use Within |
Final Diluent |
Median Concentration (S3)a |
|
| Capreomycin | Water | — | — | 1 mg/mL | 7 days | Water | 100 µg/mL |
| Chloramphenicol | Alcohol | 10 mg/mL | Water | 1 mg/mL | 30 days | Water | 2.5 µg/mL |
| Chlortetracycline | 0.01 N hydrochloric acid | — | — | 1 mg/mL | 4 days | Water | 0.06 µg/mL |
| Dihydrostreptomycinb | Water | — | — | 1 mg/mL | 30 days | Water | 30 µg/mL |
| Gramicidin | Alcohol | — | — | 1 mg/mL | 30 days | Alcohol | 0.04 µg/mL |
| Neomycinb,d | B.3c | — | — | 100 µg/mL | 14 days | B.3c | 1.0 µg/mL |
| Oxytetracycline | 0.1 N hydrochloric acid | — | — | 1 mg/mL | 4 days | Water | 0.24 µg/mL |
| Tetracycline | 0.1 N hydrochloric acid | — | — | 1 mg/mL | 1 day | Water | 0.24 µg/mL |
| Thiostrepton | Dimethyl sulfoxide | — | — | 1 U/mL | Same day | Dimethyl sulfoxide |
0.80 U/mL |
| Troleandomycin | Isopropyl alcohol and water (4:1) | — | — | 1 mg/mL | Same day | Water | 25 µg/mL |
| Tylosin | Methanol | 10 mg/mL | B.16c | 1 mg/mL | 30 days | Methanol and B.3c (1:1) | 4 µg/mL |
|
a
µg in this column refers to µg of activity.
b
The cylinder-plate assay can be used as an alternative procedure.
c
The letter B refers to buffer. See Media and Solutions, Buffers for a description of each buffer listed in this table.
d
Dilute the 100-µg/mL stock solution with Buffer B.3 to obtain a solution having a concentration equivalent to 25 µg/mL of neomycin.To separate 50-mL volumetric flasks add 1.39, 1.67, 2.00, 2.40, and 2.88 mL of this solution. Add 5.0 mL of 0.01 N hydrochloric acid to each flask, dilute with Buffer B.3 to volume, and mix to obtain solutions having concentrations of 0.69, 0.83, 1.0, 1.2, and 1.44 µg/mL of neomycin. Use these solutions to prepare the standard response line.
|
|||||||
Inocula:
Suspend the test organism from a freshly grown slant or culture in 3 mL of sterile saline TS. Glass beads can be used to facilitate the suspension. Enterococcus hirae (ATCC 10541) and Staphylococcus aureus (ATCC 9144) are grown in a liquid medium, not on agar. Spread the saline suspension onto the surface of two or more agar plates (covering the entire surface) or onto the surface of a Roux bottle containing 250 mL of the specified medium (see Table 8). Incubate at the time and temperature specified in Table 8, or until growth is apparent.
After incubation, harvest the organism from the plates or Roux bottle with approximately 50 mL of sterile saline TS, using a sterile bent glass rod or sterile glass beads. Pipet the suspension into a sterile glass bottle. This is the harvest suspension.
Determine during method verification the quantity of harvest suspension that will be used as the inoculum, starting with the volume suggested in Table 8. Prepare also an extra S3 as a test of growth. Incubate the trial tests for the times indicated in Table 11. Adjust the quantity of inoculum daily, if necessary, to obtain the optimum concentration–response relationship from the amount of growth of the test organism in the assay tubes. At the completion of the specified incubation periods, tubes containing the median concentration of the standard should have absorbance values as specified in Table 9. Determine the exact duration of incubation by observing the growth in the reference concentration (median concentration) of the standard (S3).
Table 8
| Antibiotic | Test Organism | ATCCa
Number |
Incubation Conditions | Suggested Inoculum Composition |
|||
|---|---|---|---|---|---|---|---|
| Mediumb | Temperature ( |
Time | Mediumb | Amount (mL/100 mL) |
|||
| Capreomycin | Klebsiella pneumoniae | 10031 | 1 | 36–37.5 | 16–24 h | 3 | 0.05 |
| Chloramphenicol | Escherichia coli | 10536 | 1 | 32–35 | 24 h | 3 | 0.7 |
| Chlortetracycline | Staphylococcus aureus | 29737 | 1 | 32–35 | 24 h | 3 | 0.1 |
| Dihydrostreptomycin | Klebsiella pneumoniae | 10031 | 1 | 36–37.5 | 16–24 h | 3 | 0.1 |
| Gramicidin | Enterococcus hirae | 10541 | 3 | 36–37.5 | 16–18 h | 3 | 1.0 |
| Neomycin | Klebsiella pneumoniae | 10031 | 1 | 36–37.5 | 16–24 h | 39 | 2 |
| Oxytetracycline | Staphylococcus aureus | 29737 | 1 | 32–35 | 24 h | 3 | 0.1 |
| Tetracycline | Staphylococcus aureus | 29737 | 1 | 32–35 | 24 h | 3 | 0.1 |
| Thiostrepton | Enterococcus hirae | 10541 | 40 | 36–37.5 | 18–24 h | 41 | 0.2 |
| Troleandomycin | Klebsiella pneumoniae | 10031 | 1 | 36–37.5 | 16–24 h | 3 | 0.1 |
| Tylosin | Staphylococcus aureus | 9144 | 3 | 35–39 | 16–18 h | 39 | 2–3 |
|
a
American Type Culture Collection, 10801 University Boulevard, Manassas VA 20110-2209 (http://www.atcc.org)
b
See Media and Solutions, Media.
|
|||||||
Table 9
| Antibiotic | Absorbance, NLT (a.u.) |
|---|---|
| Capreomycin | 0.4 |
| Chlortetracycline | 0.35 |
| Gramicidin | 0.35 |
| Tetracycline | 0.35 |
| All others | 0.3 |
Analysis:
On the day of the assay, prepare the necessary concentration of antibiotic by dilution of stock solutions of the standard and of each sample as specified under Standard solutions and Sample solutions. Prepare five test levels, each in triplicate, of the standard (S1–S5) and a single test level (U3), also in triplicate, of up to 20 samples corresponding to S3 (median concentration) of the standard.
Table 10
| Antibiotic | Volume of Test Dilution (mL) |
Volume of Inoculum (mL) |
|---|---|---|
| Gramicidin | 0.10 | 9.0 |
| Thiostrepton | 0.10 | 10.0 |
| Tylosin | 0.10 | 9.0 |
| All others | 1.0 | 9.0 |
Place the tubes in test tube racks or other carriers. Include in each rack 1–2 control tubes containing 1 mL of the inoculum medium (see Table 8) but no antibiotic. Add the volumes of the standard and sample test dilutions as indicated in Table 10. Randomly distribute one complete set, including the controls, in a tube rack. Add the volume of inoculum specified in Table 10 to each tube in the rack in turn, and place the completed rack immediately in an incubator or a water bath maintained at the temperature specified in Table 8 and for the time specified in Table 11.
Table 11
| Antibiotic | Incubation Time (h) |
|---|---|
| Capreomycin | 3–4 |
| Chloramphenicol | 3–4 |
| Cycloserine | 3–4 |
| Dihydrostreptomycin | 3–4 |
| Streptomycin | 3–4 |
| Troleandomycin | 3–4 |
| Tylosin | 3–5 |
| All others | 4–5 |
After incubation, immediately inhibit the growth of the organism by adding 0.5 mL of dilute formaldehyde to each tube, except for tylosin. For tylosin, heat the rack in a water bath at 80–90 for 2–6 min or in a steam bath for 5–10 min, and bring to room temperature. Read absorbance or transmittance at 530 or 580 nm, analyzing one rack at a time.
Media and Solutions
The media required for the preparation of test organism inocula are made from the ingredients listed herein. Minor modifications of the individual ingredients are acceptable; and reconstituted dehydrated media can be substituted, provided that the resulting media possess equal or better growth-promoting properties and give a similar standard curve response.
Media:
Dissolve the ingredients in water to make 1 L, and adjust the solutions with either 1 N sodium hydroxide or 1 N hydrochloric acid as required, so that after steam sterilization the pH is as specified.
Medium 1
| Peptone | 6.0 g |
| Pancreatic digest of casein | 4.0 g |
| Yeast extract | 3.0 g |
| Beef extract | 1.5 g |
| Dextrose | 1.0 g |
| Agar | 15.0 g |
| Water | 1000 mL |
| pH after sterilization | 6.6 ± 0.1 |
Medium 2
| Peptone | 6.0 g |
| Yeast extract | 3.0 g |
| Beef extract | 1.5 g |
| Agar | 15.0 g |
| Water | 1000 mL |
| pH after sterilization | 6.6 ± 0.1 |
Medium 3
| Peptone | 5.0 g |
| Yeast extract | 1.5 g |
| Beef extract | 1.5 g |
| Sodium chloride | 3.5 g |
| Dextrose | 1.0 g |
| Dibasic potassium phosphate | 3.68 g |
| Monobasic potassium phosphate | 1.32 g |
| Water | 1000 mL |
| pH after sterilization | 7.0 ± 0.05 |
Medium 4
| Peptone | 6.0 g |
| Yeast extract | 3.0 g |
| Beef extract | 1.5 g |
| Dextrose | 1.0 g |
| Agar | 15.0 g |
| Water | 1000 mL |
| pH after sterilization | 6.6 ± 0.1 |
Medium 5
| Peptone | 6.0 g |
| Yeast extract | 3.0 g |
| Beef extract | 1.5 g |
| Agar | 15.0 g |
| Water | 1000 mL |
| pH after sterilization | 7.9 ± 0.1 |
Medium 8
| Peptone | 6.0 g |
| Yeast extract | 3.0 g |
| Beef extract | 1.5 g |
| Agar | 15.0 g |
| Water | 1000 mL |
| pH after sterilization | 5.9 ± 0.1 |
Medium 9
| Pancreatic digest of casein | 17.0 g |
| Papaic digest of soybean | 3.0 g |
| Sodium chloride | 5.0 g |
| Dibasic potassium phosphate | 2.5 g |
| Dextrose | 2.5 g |
| Agar | 20.0 g |
| Water | 1000 mL |
| pH after sterilization | 7.2 ± 0.1 |
Medium 10
| Pancreatic digest of casein | 17.0 g |
| Papaic digest of soybean | 3.0 g |
| Sodium chloride | 5.0 g |
| Dibasic potassium phosphate | 2.5 g |
| Dextrose | 2.5 g |
| Agar | 12.0 g |
| Water | 1000 mL |
| Polysorbate 80 (added after boiling the medium to dissolve the agar) | 10 mL |
| pH after sterilization | 7.2 ± 0.1 |
Medium 11
| Peptone | 6.0 g |
| Pancreatic digest of casein | 4.0 g |
| Yeast extract | 3.0 g |
| Beef extract | 1.5 g |
| Dextrose | 1.0 g |
| Agar | 15.0 g |
| Water | 1000 mL |
| pH after sterilization | 8.3 ± 0.1 |
Medium 13
| Peptone | 10.0 g |
| Dextrose | 20.0 g |
| Water | 1000 mL |
| pH after sterilization | 5.6 ± 0.1 |
Medium 19
| Peptone | 9.4 g |
| Yeast extract | 4.7 g |
| Beef extract | 2.4 g |
| Sodium chloride | 10.0 g |
| Dextrose | 10.0 g |
| Agar | 23.5 g |
| Water | 1000 mL |
| pH after sterilization | 6.1 ± 0.1 |
Medium 32
| Peptone | 6.0 g |
| Pancreatic digest of casein | 4.0 g |
| Yeast extract | 3.0 g |
| Beef extract | 1.5 g |
| Manganese sulfate | 0.3 g |
| Dextrose | 1.0 g |
| Agar | 15.0 g |
| Water | 1000 mL |
| pH after sterilization | 6.6 ± 0.1 |
Medium 34
| Glycerol | 10.0 g |
| Peptone | 10.0 g |
| Beef extract | 10.0 g |
| Sodium chloride | 3.0 g |
| Water | 1000 mL |
| pH after sterilization | 7.0 ± 0.1 |
Medium 35
| Glycerol | 10.0 g |
| Peptone | 10.0 g |
| Beef extract | 10.0 g |
| Sodium chloride | 3.0 g |
| Agar | 17.0 g |
| Water | 1000 mL |
| pH after sterilization | 7.0 ± 0.1 |
Medium 36
| Pancreatic digest of casein | 15.0 g |
| Papaic digest of soybean | 5.0 g |
| Sodium chloride | 5.0 g |
| Agar | 15.0 g |
| Water | 1000 mL |
| pH after sterilization | 7.3 ± 0.1 |
Medium 39
| Peptone | 5.0 g |
| Yeast extract | 1.5 g |
| Beef extract | 1.5 g |
| Sodium chloride | 3.5 g |
| Dextrose | 1.0 g |
| Dibasic potassium phosphate | 3.68 g |
| Monobasic potassium phosphate | 1.32 g |
| Water | 1000 mL |
| pH after sterilization | 7.9 ± 0.1 |
Medium 40
| Yeast extract | 20.0 g |
| Polypeptone | 5.0 g |
| Dextrose | 10.0 g |
| Monobasic potassium phosphate | 2.0 g |
| Polysorbate 80 | 0.1 g |
| Agar | 10.0 g |
| Water | 1000 mL |
| pH after sterilization | 6.7 ± 0.2 |
Medium 41
| Pancreatic digest of casein | 9.0 g |
| Dextrose | 20.0 g |
| Yeast extract | 5.0 g |
| Sodium citrate | 10.0 g |
| Monobasic potassium phosphate | 1.0 g |
| Dibasic potassium phosphate | 1.0 g |
| Water | 1000 mL |
| pH after sterilization | 6.8 ± 0.1 |
Solutions
Buffers:
Prepare as directed in Table 12, or by other suitable means. The buffers are sterilized after preparation; the pH specified in each case is the pH after sterilization.
Table 12. Buffers
| Buffer | Concentration of Dibasic Potassium Phosphate (g/L) |
Concentration of Monobasic Potassium Phosphate (g/L) |
Volume of 10 N Potassium Hydroxide (mL) |
pH after Sterilizationa |
|---|---|---|---|---|
| Buffer B.1 (1%, pH 6.0) | 2 | 8 | — | 6.0 ± 0.05 |
| Buffer B.3 (0.1 M, pH 8.0) | 16.73 | 0.523 | — | 8.0 ± 0.1 |
| Buffer B.4 (0.1 M, pH 4.5) | — | 13.61 | — | 4.5 ± 0.05 |
| Buffer B.6 (10%, pH 6.0) | 20 | 80 | — | 6.0 ± 0.05 |
| Buffer B.10 (0.2 M, pH 10.5) | 35 | — | 2 | 10.5 ± 0.1 |
| Buffer B.16 (0.1 M, pH 7.0) | 13.6 | 4 | — | 7.0 ± 0.2 |
|
a
Adjust the pH with 18 N phosphoric acid or 10 N potassium hydroxide.
|
||||
Other solutions:
See Reagents, Indicators, and Solutions.
Water:
Use Purified Water.
Saline:
Use saline TS.
Dilute formaldehyde:
Formaldehyde solution and water (1:3)
Calculations
Introduction:
Antibiotic potency is calculated by interpolation from a standard curve using a log-transformed straight-line method with a least-squares fitting procedure (see below for calculation details). The analyst must consider three essential concepts in interpreting antibiotic potency results:
- Biological concentration–response relationships generally are not linear. The antibiotic potency method allows fitting the data to a straight line by evaluating a narrow concentration range where the results approach linearity. The assay results can be considered valid only if the computed potency is 80%–125% of that assumed in preparing the sample stock solution. When the calculated potency value falls outside 80%–125%, the result for the sample may fall outside the narrow concentration range where linearity has been established. In such a case, adjust the assumed potency of the sample accordingly, and repeat the assay to obtain a valid result.
- The most effective means of reducing the variability of the reportable value (the geometric mean potency across runs and replicates) is through independent runs of the assay procedure. The combined result of a series of smaller, independent assays spread over a number of days is a more reliable estimate of potency than that from a single large assay with the same total number of plates or tubes. Three or more independent assays are required for antibiotic potency determinations.
- The number of assays needed in order to obtain a reliable estimate of antibiotic potency depends on the required specification range and the assay variability. The confidence limit calculation described below is determined from several estimated log potencies that are approximately equal in precision. If the value calculated for the width of the confidence interval, W, is too wide, no useful decision can be made about whether the potency meets its specification.
The laboratory should predetermine in its standard operating procedures a maximum acceptable value for the confidence interval width. This maximum value should be determined during development and confirmed during validation or verification. If the calculated confidence interval width exceeds this limit, the analyst must perform additional independent potency determinations to meet the limit requirement. Note that the decision to perform additional determinations does not depend on the estimated potency but only on the uncertainty in that estimate as determined by the confidence interval width. Assay variability has a greater impact on the calculated confidence limit than does the number of independent potency determinations. As a result, the analyst should first consider decreasing variability to the extent possible before conducting potency determinations.
The following sections describe the calculations for determining antibiotic potency as well as for performing the confidence limit calculation. Methods for calculating standard error are also shown in order to allow estimates of assay variance. Where logarithms are used, any base log is acceptable. Appendix 1 provides formulas for hand calculations applicable when the concentrations are equally spaced in the log scale. Alternative statistical methods may be used if appropriately validated.
Cylinder-plate assay:
This section details analysis of the sample data and determination of the potency of an unknown, using the cylinder-plate assay.
Sample data:
Table 13 shows the data from one assay that will be used as an example throughout this section. For each of the 12 plates, zones 1, 3, and 5 are the reference concentration and the other three zones are for one of the other four concentrations, as shown. Other columns are needed for calculations and are explained below.
Step 1:
Perform initial calculations and variability suitability check.
For each set of three plates, average the nine reference values and average the nine standard values.
Example
(see Table 13)
15.867 = X (16.1, 15.6, …, 15.8)
14.167 = X(14.6, 14.1, …, 14.8)
For each set of three plates determine the standard deviation of the nine reference values and the standard deviation of the nine standard values. For each standard deviation, determine the corresponding relative standard deviation.
Example
(see Table 13)
0.200 = 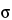 (16.1, …, 15.8)
(16.1, …, 15.8)
1.3% = (0.200/15.867) × 100
0.324 = (14.6, …, 14.1)
2.3% = (0.324/14.167) × 100
For a variability suitability criterion, each laboratory should determine a maximum acceptable value for the relative standard deviation. If any of the eight relative standard deviations (four for the reference and four for the standard) exceed this predetermined maximum, the assay data are not suitable and should be discarded. [Note—The suggested limit for relative standard deviation is NMT 10%. ]
Step 2:
Perform a plate-to-plate variation correction.
XC = corrected standard mean
XS = original standard mean
XR = reference mean
P = correction point
This correction is applied to convert the average zone measurement obtained for each concentration to the value it would be if the average reference concentration measurement for that set of three replicate plates were the same as the value of the correction point:
XC = XS (XR P)
XC = corrected standard mean
XS = original standard mean
XR = reference mean
P = correction point
Example:
For the first set of three plates in Table 13 (S1), the correction is:
14.022 = 14.167 (15.867 15.722) = 14.167 0.145
Step 3:
Determine the standard curve line.
Generate the standard curve line by plotting the corrected zone measurements versus the log of the standard concentration values. Calculate the equation of the standard curve line by performing a standard unweighted linear regression on these values, using appropriate software or the manual calculations of Appendix 1. [Note—Use either the natural log or the base 10 log to plot the standard curve and determine the regression equation; both provide the same final test result. ] Each laboratory should determine a minimum value of the coefficient of determination (%R2) for an acceptable regression. The regression is acceptable only if the obtained %R2 exceeds this predetermined value. [Note—The suggested limit for the percentage coefficient of determination is NLT 95%. ]
Table 13. Sample Data (Cylinder-Plate Assay)
| Standard | Concentration (U/mL) |
Plate replicate |
Reference (S3) | Sample | Corrected Mean (mm) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Zone 1 (mm) | Zone 3 (mm) |
Zone 5 (mm) |
Mean (mm) |
SD | %RSD | Zone 2 (mm) |
Zone 4 (mm) |
Zone 6 (mm) |
Mean (mm) |
SD | %RSD | ||||
| S1 | 3.20 | 1 | 16.1 | 15.6 | 15.8 | 15.867 | 0.200 | 1.3 | 14.6 | 14.1 | 13.5 | 14.167 | 0.324 | 2.3 | 14.022 |
| 2 | 16.0 | 15.9 | 16.2 | 14.5 | 14.1 | 14.4 | |||||||||
| 3 | 15.7 | 15.7 | 15.8 | 14.0 | 14.2 | 14.1 | |||||||||
| S2 | 4.00 | 1 | 15.8 | 15.6 | 15.5 | 15.567 | 0.158 | 1.0 | 14.7 | 15.1 | 14.8 | 14.833 | 0.265 | 1.8 | 14.989 |
| 2 | 15.7 | 15.5 | 15.6 | 14.7 | 14.9 | 15.2 | |||||||||
| 3 | 15.7 | 15.4 | 15.3 | 14.8 | 15.0 | 14.3 | |||||||||
| S4 | 6.25 | 1 | 15.6 | 15.8 | 16.0 | 15.789 | 0.169 | 1.1 | 16.6 | 16.8 | 16.3 | 16.578 | 0.233 | 1.4 | 16.511 |
| 2 | 15.8 | 15.6 | 15.7 | 16.6 | 16.5 | 16.2 | |||||||||
| 3 | 16.1 | 15.7 | 15.8 | 16.9 | 16.5 | 16.8 | |||||||||
| S5 | 7.8125 | 1 | 15.6 | 15.6 | 15.5 | 15.667 | 0.141 | 0.9 | 17.3 | 17.0 | 17.0 | 17.167 | 0.224 | 1.3 | 17.222 |
| 2 | 15.6 | 15.7 | 15.5 | 17.3 | 17.4 | 17.2 | |||||||||
| 3 | 15.9 | 15.8 | 15.8 | 17.3 | 17.3 | 16.7 | |||||||||
| 15.722a | |||||||||||||||
| U3 | unknown | 1 | 15.7 | 15.8 | 15.7 | 15.678 | 0.179 | 1.1 | 15.3 | 15.8 | 15.7 | 15.478 | 0.307 | 2.0 | 15.522 |
| 2 | 15.9 | 15.7 | 15.7 | 15.8 | 15.8 | 15.5 | |||||||||
| 3 | 15.5 | 15.8 | 15.3 | 15.2 | 15.1 | 15.1 | |||||||||
|
a
This is the value of the overall reference mean, referred to as the “correction point” below.
|
|||||||||||||||
Linear regression results
 %R2 = 99.7
%R2 = 99.7
Standard curve line:
Z = corrected zone measurement
C = concentration
Z = [3.551 × ln(C)] + 9.978
Z = corrected zone measurement
C = concentration
Sample potency determination:
To estimate the potency of the unknown sample, average the zone measurements of the standard and the zone measurements of the sample on the three plates used. Correct for plate-to-plate variation using the correction point determined above to obtain a corrected average for the unknown, U. [Note—An acceptable alternative to using the correction point is to correct using the value on the estimated regression line corresponding to the log concentration of S3. ] Use the corrected average zone measurement in the equation of the standard curve line to determine the log concentration of the sample, LU, by:
a = intercept of the regression line
b = slope of the regression line
LU = (U a)/b
a = intercept of the regression line
b = slope of the regression line
To obtain the potency of the unknown, take the antilog of LU and multiply the result by any applicable dilution factor. This value can also be expressed as a percentage of the reference concentration value.
Example:
Corrected sample zone measurement (Table 13) = 15.522
Natural log of the sample concentration:
LU = (15.522 9.978)/3.551 = 1.561
Sample concentration:
CU = e1.561 = 4.765
Percentage of reference concentration:
Result = (4.765/5.000) × 100 = 95.3%
Turbidimetric assay:
This section details analysis of the sample data and determination of the potency of an unknown using the turbidimetric assay. The method assumes that the tubes are randomly distributed within the heat block or other temperature control device. If the device has a temperature profile that is not uniform, a randomized blocks design is preferred. In such a design, the rack is divided into areas (blocks) of relatively uniform temperature and at least one tube of each Standard concentration and of each unknown is placed in each area. The data analysis of a randomized block design is different from the following.
Sample data:
Table 15 shows the data from one assay that will be used for an example throughout this section. Other columns are needed for calculations and are explained below.
Table 15. Sample Data (Turbidimetric Assay)
| Standard | Concentration (µg/mL) |
Replicate | Absorbance (a.u.) |
Average (a.u.) |
Standard Deviation |
|---|---|---|---|---|---|
| S1 | 64 | 1 | 0.8545 | 0.8487 | 0.0062 |
| 2 | 0.8422 | ||||
| 3 | 0.8495 | ||||
| S2 | 80 | 1 | 0.8142 | 0.8269 | 0.0125 |
| 2 | 0.8273 | ||||
| 3 | 0.8392 | ||||
| S3 | 100 | 1 | 0.6284 | 0.6931 | 0.0640 |
| 2 | 0.6947 | ||||
| 3 | 0.7563 | ||||
| S4 | 125 | 1 | 0.6933 | 0.6827 | 0.0119 |
| 2 | 0.6850 | ||||
| 3 | 0.6699 | ||||
| S5 | 156 | 1 | 0.5299 | 0.5465 | 0.0272 |
| 2 | 0.5779 | ||||
| 3 | 0.5316 | ||||
| U3 | unknown | 1 | 0.7130 | 0.7430 | 0.0460 |
| 2 | 0.7960 | ||||
| 3 | 0.7201 | ||||
Step 1:
Perform initial calculations and variability suitability check.
For each concentration, determine the standard deviation of the three readings and a combined standard deviation for all the concentrations.
For each concentration (including the sample), average the three absorbance values.
Example:
See S1 in Table 15.
0.0125 = SD(0.8545, 0.8422, 0.8495)
The combined value is calculated by taking the square root of the average of the five variances:
0.0325 = {[(0.0062)2 + (0.0125)2 + (0.0640)2 + (0.0119)2 + (0.0272)2]/5}1/2
For a variability suitability criterion, each laboratory should determine a maximum acceptable combined standard deviation. If the combined standard deviation exceeds this predetermined maximum, the assay data are not suitable and should be discarded. [Note—The suggested limit for the combined standard deviation is NMT 10% of the average absorbance value across the five concentrations. ] If the number of replicates per concentration is at least five, then a relative standard deviation can be computed for each concentration after checking for outliers and compared to a maximum acceptable relative standard deviation. [Note—The suggested limit for the relative standard deviation is NMT 10%. ]
Step 2:
Determine the standard curve line.
Generate the standard curve line by plotting the average absorbance values versus the log of the standard concentration values. Calculate the equation of the standard curve line by performing an unweighted linear regression on these values using appropriate software or the manual calculations of Appendix 1. [Note—Use either the natural log or the base 10 log to plot the standard curve and determine the regression equation; both provide the same final test result. ] Each laboratory should determine a minimum value of the percentage coefficient of determination (%R2) for an acceptable regression. The regression is acceptable only if the %R2 value obtained exceeds this predetermined value. [Note—The suggested limit for the percentage coefficient of determination is NLT 90%. ]
Linear regression results
%R2 = 93.0%
Standard curve line:
Absorbance = 2.2665 [0.7735 × log10(concentration)]
Sample potency determination:
To estimate the potency of the unknown sample, average the three absorbance measurements to obtain an average for the unknown, U. Use this average measurement in the equation of the standard curve line to determine the log concentration of the unknown sample, LU, by:
a = intercept of the regression line
b = slope of the regression line
LU = (U a)/b
a = intercept of the regression line
b = slope of the regression line
To obtain the potency of the unknown, take the antilog of LU and multiply the result by any applicable dilution factor. This value can also be expressed as a percentage of the reference concentration value.
Example:
Average sample absorbance (Table 15) = 0.7430.
CU = concentration of the sample
log10(CU) = (0.7430 2.2665)/( 0.7735) = 1.9696
CU = 101.9696 = 93.2
Percentage of reference concentration = (93.2/100.0) × 100 = 93.2%
CU = concentration of the sample
Confidence limits and combination of assays calculations:
Because of interassay variability, three or more independent determinations are required for a reliable estimate of the sample potency. For each independent determination, start with separately prepared stock solutions and test dilutions of both the Standard and the sample, and repeat the assay of a given sample on a different day.
M = average
SD = standard deviation
N = number of assays
t (0.05, N1) = the two-sided 5% point of a Student's t-distribution with N1 degrees of freedom
W = half-width of the confidence interval
Given a set of at least three determinations of the unknown potency, use the method of Appendix 2 to check for any outlier values. This determination should be done in the log scale.
To obtain a combined estimate of the unknown potency, calculate the average, M, and the standard deviation of the accepted log potencies. [Note—Use either the natural log or the base 10 log. ] Determine the confidence interval for the potency as follows:
antilog[M t(0.05, N 1) × SD/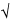 N], antilog[M + t(0.05, N 1) × SD/N]
N], antilog[M + t(0.05, N 1) × SD/N]
M = average
SD = standard deviation
N = number of assays
t (0.05, N
Note—The t value is available in spreadsheets, statistics texts, and statistics software.
W = antilog{[t(0.05, N 1) × SD/N]}
W = half-width of the confidence interval
Compare the half-width of the confidence interval to a predetermined maximum acceptable value. If the half-width is larger than the acceptance limit, continue with additional assays.
Example:
Suppose the sample is assayed four times, with potency results in the natural log scale of 1.561, 1.444, 1.517, and 1.535. Then:
N = 4
M = X(1.561, 1.444, 1.517, 1.535) = 1.514
SD = (1.561, 1.444, 1.517, 1.535) = 0.050
t = 3.182
The confidence interval in the log scale is
1.514 ± (3.182 × 0.050/4) = (1.434, 1.594)
Taking antilogs, the estimated potency is
e1.514 = 4.546
with a 95% confidence interval for the potency of e1.434, e 1.594 = (4.197, 4.924).
The confidence interval half-width to compare to an acceptance value is the ratio 4.924/4.546 = 1.083.
Appendix 1. Formulas for Manual Calculations of Regression and Sample Concentration
If the concentrations are equally spaced in the logarithmic scale, the calculations can be performed using the following formula. Let:
Sk = mean corrected zone measurement (cylinder-plate assay) or average absorbance value (turbidimetric assay) for standard set k
k = 1, 2, 3, 4, 5
S = mean of the five Sk values
Lk = logarithm of the kth concentration. [Note—Use either the natural log or the base 10 log. Slope of the regression line is calculated by: ]
b = (Yhigh Ylow)/(Xhigh Xlow)
Yhigh = 1/5(3S5 + 2S4 + S3 S1)
Ylow = 1/5(3S1 + 2S2 + S3 S5)
Xhigh = L5
Xlow = L1
Combine and simplify to:
b = (4S5 + 2S4 2S2 4S1)/[5(L5 L1)]
The log of the concentration of the sample is found using:
LU = Lreference + [(U – S)/b]
For example, using the data for the cylinder-plate assay in Table 13 and natural logarithms:
b = [(4 × 17.222) + (2 × 16.511) (2 × 14.989) (4 × 14.020)]/{5[ln(7.81)] ln(3.2)} = 3.551
S = (14.020 + 14.989 + 15.722 + 16.511 + 17.222)/5 = 15.693
Natural log of sample concentration = ln(5) + [(15.522 15.693)/3.551] = 1.561
Sample concentration = e1.561 = 4.765
Appendix 2. Procedure for Checking for Outliers; Rejection of Outlying or Aberrant Measurements
A measurement that is clearly questionable because of a failure in the assay procedure should be rejected, whether it is discovered during the measuring or tabulation procedure. The arbitrary rejection or retention of an apparently aberrant measurement can be a serious source of bias. In general, the rejection of measurements solely on the basis of their relative magnitudes is a procedure that should be used sparingly.
Each suspected potency measurement, or outlier, may be tested against the following criterion. This criterion is based on the variation within a single group of supposedly equivalent measurements from a normal distribution. On average, it will reject a valid observation once in 25 trials or once in 50 trials. Designate the measurements in order of magnitude from y1 to yN, where y1 is the candidate outlier, and N is the number of measurements in the group. Compute the relative gap by using Table A2-1, Test for Outlier Measurements, and the formulas below:
When N = 3 to 7:
G1 = (y2 y1)/(yN y1)
When N = 8 to 10:
G2 = (y2 y1)/(yN 1 y1)
When N = 11 to 13:
G3 = (y3 y1)/(yN 1 y1)
If G1, G2, or G3, as appropriate, exceeds the critical value in Table A2-1, Test for Outlier Measurements, for the observed N, there is a statistical basis for omitting the outlier measurement(s).
Table A2-1. Test for Outlier Measurements
| In samples from a normal population, gaps equal to or larger than the following values of G1, G2, and G3 occur with a probability P = 0.01, when outlier measurements can occur only at one end; or with P = 0.02, when they may occur at either end. | |||||
|---|---|---|---|---|---|
| N | 3 | 4 | 5 | 6 | 7 |
| G1 | 0.987 | 0.889 | 0.781 | 0.698 | 0.637 |
| N | 8 | 9 | 10 | ||
| G2 | 0.681 | 0.634 | 0.597 | ||
| N | 11 | 12 | 13 | ||
| G3 | 0.674 | 0.643 | 0.617 | ||
Example:
Estimated potencies of sample in log scale = 1.561, 1.444, 1.517, 1.535.
Check lowest potency for outlier:
G1 = (1.517 1.444)/(1.561 1.444) = 0.624 < 0.889
Therefore 1.444 is not an outlier.
Check highest potency for outlier:
G1 = (1.561 1.535)/(1.561 1.444) = 0.222 < 0.889
Therefore 1.561 is not an outlier.
Outlier potencies should be marked as outlier values and excluded from the assay calculations. NMT one potency can be excluded as an outlier. USP35
USP35
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Ahalya Wise, M.S.
Senior Scientific Liaison 1-301-816-8161 |
(GCM2010) General Chapters - Microbiology |
USP35–NF30 Page 74
Pharmacopeial Forum: Volume No. 36(5) Page 1239