INTRODUCTION
The method is based on the ISO standards 13320-1(1999) and 9276-1(1998).
This general chapter has been harmonized with the corresponding texts of the European Pharmacopoeia and/or the Japanese Pharmacopoeia.
The laser light diffraction technique used for the determination of particle-size distribution is based on the analysis of the diffraction pattern produced when particles are exposed to a beam of monochromatic light. Historically, the early laser diffraction instruments only used scattering at small angles. However, the technique has since been broadened to include laser light scattering in a wider angular range and application of the Mie theory, in addition to the Fraunhofer approximation and anomalous diffraction.
The technique cannot distinguish between scattering by single particles and scattering by clusters of primary particles, i.e., by agglomerates or aggregates. As most particulate samples contain agglomerates or aggregates and as the focus of interest is generally on the size distribution of primary particles, the clusters are usually dispersed into primary particles before measurement.
For nonspherical particles, an equivalent sphere-size distribution is obtained because the technique assumes spherical particles in its optical model. The resulting particle-size distribution may differ from those obtained by methods based on other physical principles (e.g., sedimentation, sieving).
This chapter provides guidance for the measurement of size distributions of particles in different dispersed systems (e.g., powders, sprays, aerosols, suspensions, emulsions, and gas bubbles in liquids), through analysis of their angular light-scattering patterns. It does not address specific requirements of particle-size measurement of specific products.
PRINCIPLE
A representative sample, dispersed at an adequate concentration in a suitable liquid or gas, is passed through a beam of monochromatic light, usually a laser. The light scattered by the particles at various angles is measured by a multi-element detector. Numerical values representing the scattering pattern are then recorded for subsequent analysis. These scattering pattern values are then transformed, using an appropriate optical model and mathematical procedure, to yield the proportion of total volume to a discrete number of size classes, forming a volumetric particle-size distribution.
INSTRUMENT
The instrument is located in an environment where it is not affected by electrical noise, mechanical vibrations, temperature fluctuations, humidity, or direct bright light.
An example of a setup of a laser light diffraction instrument is given in Figure 1. Other equipment may be used.
+'&img=../../images/v35300/c429-f1.gif&casNumber=&usp=35&nf=30&chemicalStructureImage=false&ID='+parent.myID,600,500);)
Figure 1. Example of a set-up of a laser light diffraction instrument.
The instrument comprises a laser light source, beam processing optics, a sample measurement region (or cell), a Fourier lens, and a multi-element detector for measuring the scattered light pattern. A data system is also required for deconvolution of the scattering data into a volumetric size distribution and associated data analysis and reporting.
The particles can enter the laser beam in two positions. In the conventional case the particles enter the parallel beam before the collecting lens and within its working distance. In so-called reversed Fourier optics the particles enter behind the collecting lens and thus in a converging beam. The advantage of the conventional setup is that a reasonable path length for the sample is allowed within the working distance of the lens. The second setup allows only small path lengths but enables measurement of scattered light at larger angles, which is useful when submicron particles are present.
The interaction of the incident light beam and the ensemble of dispersed particles results in a scattering pattern with different light intensities at various angles. The total angular intensity distribution, consisting of both direct and scattered light, is then focused onto a multi-element detector by a lens or a series of lenses. These lenses create a scattering pattern that, within limits, does not depend on the location of the particles in the light beam. Hence, the continuous angular intensity distribution is converted into a discrete spatial intensity distribution on a set of detector elements.
It is assumed that the measured scattering pattern of the particle ensemble is identical to the sum of the patterns from all individual single scattering particles presented in random relative positions. Note that only a limited angular range of scattered light is collected by the lens(es) and, therefore, by the detector.
DEVELOPMENT OF THE METHOD
The measurement of particle size by laser diffraction can give reproducible data, even in the submicron region, provided the instrument used and the sample tested are carefully controlled to limit variability of the test conditions (e.g., dispersion medium, method of preparation of the sample dispersion).
Traditionally, the measurement of particle size using laser diffraction has been limited to particles in the range of approximately 0.1 µm to 3 mm. Because of recent advances in lens and equipment design, newer instruments are routinely capable of exceeding this range. With the validation report, the user demonstrates the applicability of the method for its intended use.
Sampling
The sampling technique must be adequate to obtain a representative sample of a suitable volume for the particle-size measurement. Sample splitting techniques such as rotating riffler or the cone and quartering method may be applied.
Evaluation of the Dispersion Procedure
The sample to be analyzed is inspected, visually or with the aid of a microscope, to estimate its size range and particle shape. The dispersion procedure must be adjusted to the purpose of the measurement. The purpose may be such that it is preferable to deagglomerate clusters into primary particles as far as possible, or it may be desirable to retain clusters as intact as possible. In this sense, the particles of interest may be either primary particles or clusters.
For the development of a method, it is highly advisable to check that comminution of the particles does not occur and, conversely, that dispersion of particles or clusters is satisfactory. This can usually be done by changing the dispersing energy and monitoring the change of the particle-size distribution. The measured size distribution must not change significantly when the sample is well dispersed and the particles are neither fragile nor soluble. Moreover, if the manufacturing process (e.g., crystallization, milling) of the material has changed, the applicability of the method must be verified (e.g., by microscopic comparison).
Sprays, aerosols, and gas bubbles in a liquid should be measured directly, provided that their concentration is adequate, because sampling or dilution generally alters the particle-size distribution.
In other cases (such as emulsions, pastes, and powders), representative samples may be dispersed in suitable liquids. Dispersing aids (wetting agents, stabilizers) and/or mechanical forces (e.g., agitation, sonication) are often applied for deagglomeration or deaggregation of clusters and stabilization of the dispersion. For these liquid dispersions, a recirculating system consisting of an optical measuring cell, a dispersion bath usually equipped with stirrer and ultrasonic elements, a pump, and tubing is most commonly used. Nonrecirculating, stirred cells are useful when only small amounts of a sample are available or when special dispersion liquids are used.
Dry powders can also be converted into aerosols through the use of suitable dry powder dispersers that apply mechanical force for deagglomeration or deaggregation. Generally, the dispersers use the energy of compressed gas or the differential pressure of a vacuum to disperse the particles to an aerosol that is blown through the measuring zone, usually into the inlet of a vacuum unit that collects the particles. However, for free-flowing, coarser particles or granules, the effect of gravity may be sufficient to disperse the particles adequately.
If the maximum particle size of the sample exceeds the measuring range of the instrument, the material that is too coarse can be removed by sieving, and the mass and percentage of removed material are reported. However, after presieving, note that the sample is no longer representative, unless proven otherwise.
Optimization of the Liquid Dispersion
Liquids, surfactants, and dispersing aids used to disperse powders must
— be transparent at the laser wavelength and practically free from air bubbles or particles;
— have a refractive index that differs from that of the test material;
— be a nonsolvent of the test material (pure liquid or prefiltered, saturated solution);
— not alter the size of the test materials (e.g., by solubility, solubility enhancement, or recrystallization effects);
— favor easy formation and stability of the dispersion;
— be compatible with the materials used in the instrument (such as O-rings, gaskets, tubing, etc.); and
— possess a suitable viscosity to facilitate recirculation, stirring, and filtration.
Surfactants and/or dispersing aids are often used to wet the particles and to stabilize the dispersion. For weak acids and weak bases, buffering of the dispersing medium at low or high pH, respectively, can assist in identifying a suitable dispersant.
A preliminary check of the dispersion quality can be performed by visual or microscopic inspection. It is also possible to take fractional samples out of a well-mixed stock dispersion. Such stock dispersions are formed by adding a liquid to the sample while mixing it with, for example, a glass rod, a spatula or a vortex mixer. Care must be taken to ensure the transfer of a representative sample and that settling of larger particles does not occur. Therefore, a sample paste is prepared or sampling is carried out quickly from a suspension maintained under agitation.
Optimization of the Gas Dispersion
For sprays and dry powder dispersions, a compressed gas free from oil, water, and particles may be used. To remove such materials from the compressed gas, a dryer with a filter can be used. Any vacuum unit should be located away from the measurement zone, so that its output does not disturb the measurement.
Determination of the Concentration Range
In order to produce an acceptable signal-to-noise ratio in the detector, the particle concentration in the dispersion must exceed a minimum level. Likewise, it must be below a maximum level in order to avoid multiple scattering. The concentration range is influenced by the width of the laser beam, the path length of the measurement zone, the optical properties of the particles, and the sensitivity of the detector elements.
In view of the above, measurements must be performed at different particle concentrations to determine the appropriate concentration range for any typical sample of material. [note—In different instruments, particle concentrations are usually represented by differently scaled and differently named numbers, e.g., obscuration, optical concentration, proportional number of total mass. ]
Determination of the Measuring Time
The time of measurement, the reading time of the detector, and the acquisition frequency is determined experimentally in accordance with the required precision. Generally, the time for measurement permits a large number of detector scans or sweeps at short time intervals.
Selection of an Appropriate Optical Model
Most instruments use either the Fraunhofer or the Mie theory, though other approximation theories are sometimes applied for calculation of the scattering matrix. The choice of the theoretical model depends on the intended application and the different assumptions (size, absorbance, refractive index, roughness, crystal orientation, mixture, etc.) made for the test material. If the refractive index values (real and imaginary parts for the used wavelength) are not exactly known, then the Fraunhofer approximation or the Mie theory with a realistic estimate of the refractive index can be used. The former has the advantages that it is simple and it does not need refractive index values; the latter usually provides less-biased particle-size distributions for small particles. For instance, if the Fraunhofer model is used for samples containing an appreciable amount of small, transparent particles, a significantly larger amount of small particles may be calculated. In order to obtain traceable results, it is essential to document the refractive index values used, because small differences in the values assumed for the real and imaginary part of the complex refractive index may cause significant differences in the resulting particle-size distributions. Small values of the imaginary part of the refractive index (about 0.01–0.1 i) are often applied to allow the correction of the absorbance for the surface roughness of the particles. It should be noted, in general, that the optical properties of the substance to be tested, as well as the structure (e.g., shape, surface roughness, and porosity) bear upon the final result.
Validation
Typically, the validity of a procedure may be assessed by the evaluation of its specificity, linearity, range, accuracy, precision, and robustness. In particle-size analysis by laser light diffraction, specificity as defined by ICH is not applicable as it is not possible to discriminate different components into a sample, nor is it possible to discriminate between agglomerates from dispersed particles unless properly complemented by microscopic techniques. Exploring a linear relationship between concentration and response, or a mathematical model for interpolation, is not applicable to this procedure. Rather than evaluating linearity, this method requires the definition of a concentration range within which the result of the measurements does not vary significantly. Concentrations below that range produce an error due to a poor signal-to-noise ratio, while concentrations above that range produce an error due to multiple scattering. The range depends mostly on the instrument hardware. Accuracy should be confirmed through an appropriate instrument qualification and comparison with microscopy, while precision may be assessed by means of a repeatability determination.
The attainable repeatability of the method mainly depends on the characteristics of the material (milled/not milled, robust/fragile, width of its size distribution, etc.), whereas the required repeatability depends on the purpose of the measurement. Mandatory limits cannot be specified in this chapter, as repeatabilities (different sample preparations) may vary appreciably from one substance to another. However, it is good practice to aim at acceptance criteria for repeatability such as % RSD 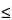 10% [n = 6] for any central value of the distribution (e.g., for x50). Values at the sides of the distribution (e.g., x10 and x90) are oriented towards less stringent acceptance criteria such as % RSD 15% [n = 6]. Below 10 µm, these values must be doubled. Robustness may be tested during the selection and optimization of the dispersion media and forces. The change of the dispersing energy may be monitored by the change in the particle-size distribution.
10% [n = 6] for any central value of the distribution (e.g., for x50). Values at the sides of the distribution (e.g., x10 and x90) are oriented towards less stringent acceptance criteria such as % RSD 15% [n = 6]. Below 10 µm, these values must be doubled. Robustness may be tested during the selection and optimization of the dispersion media and forces. The change of the dispersing energy may be monitored by the change in the particle-size distribution.
MEASUREMENT
Precautions
The instructions given in the instrument manual are followed:
— never look into the direct path of the laser beam or its reflections;
— earth all instrument components to prevent ignition of solvents or dust explosions;
— check the instrument set-up (e.g., warm-up, required measuring range and lens, appropriate working distance, position of the detector, no direct bright daylight); and
— in the case of wet dispersions, avoid air bubbles, evaporation of liquid, schlieren or other inhomogeneities in the dispersion; similarly, avoid improper mass-flow from the disperser or turbulent airflow in the case of dry dispersions; such effects can cause erroneous particle-size distributions.
Measurement of the Light Scattering of Dispersed Sample(s)
After proper alignment of the optical part of the instrument, a blank measurement of the particle-free dispersion medium must be performed using the same method as that used for the measurement of the sample. The background signal must be below an appropriate threshold. The detector data are saved in order to subtract them later from the data obtained with the sample. The sample dispersion is measured according to the developed method.
For each detector element, an average signal is calculated, sometimes together with its standard deviation. The magnitude of the signal from each detector element depends upon the detection area, the light intensity, and the quantum efficiency. The coordinates (size and position) of the detector elements together with the focal distance of the lens determine the range of scattering angles for each element. Most instruments also measure the intensity of the central (unscattered) laser beam. The ratio of the intensity of a dispersed sample to that in its absence (the blank measurement) indicates the proportion of scattered light and hence the particle concentration.
Conversion of Scattering Pattern Into Particle-Size Distribution
This deconvolution step is the inverse of the calculation of a scattering pattern for a given particle-size distribution. The assumption of spherical particle shape is particularly important as most algorithms use the mathematical solution for scattering from spherical particles. Furthermore, the measured data always contain some random and systematic errors, which may vitiate the size distributions. Several mathematical procedures have been developed for use in the available instruments. They contain some weighting of deviations between measured and calculated scattering patterns (e.g., least squares), some constraints (e.g., non-negativity for amounts of particles), and/or some smoothing of the size distribution curve.
The algorithms used are specific to each make and model of equipment, and are proprietary. The differences in the algorithms between different instruments may give rise to differences in the calculated particle-size distributions.
Replicates
The number of replicate measurements (with individual sample preparations) to be performed, depends on the required measurement precision. It is recommended to set this number in a substance-specific method.
REPORTING RESULTS
The particle-size distribution data are usually reported as cumulative undersize distribution and/or as density distribution by volume. The symbol x is used to denote the particle size, which in turn is defined as the diameter of a volume-equivalent sphere. Q3(x) denotes the volume fraction undersize at the particle size x. In a graphical representation, x is plotted on the abscissa and the dependent variable Q3 on the ordinate. Most common characteristic values are calculated from the particle-size distribution by interpolation. The particle sizes at the undersize values of 10%, 50%, and 90% (denoted as x10, x50, and x90, respectively) are frequently used. x50 is also known as the median particle size. It is recognized that the symbol d is also widely used to designate the particle size, thus the symbol x may be replaced by d.
Moreover, sufficient information must be documented about the sample, the sample preparation, the dispersion conditions, and the cell type. Because the results depend on the particular instrument, data analysis program, and optical model used, these details must also be documented.
CONTROL OF THE INSTRUMENT PERFORMANCE
Use the instrument according to the manufacturer's instructions and carry out the prescribed qualifications at an appropriate frequency, according to the use of the instrument and substances to be tested.
Calibration
Laser diffraction systems, although assuming idealized properties of the particles, are based on first principles of laser light scattering. Thus, calibration in the strict sense is not required. However, it is still necessary to confirm that the instrument is operating correctly. This can be undertaken using any certified reference material that is acceptable in industrial practice. The entire measurement procedure is examined, including sample collection, sample dispersion, sample transport through the measuring zone, measurement, and the deconvolution procedure. It is essential that the total operational procedure is fully described.
The preferred certified reference materials consist of spherical particles of a known distribution. They must be certified as to the mass-percentage size distribution by an absolute technique, if available, and used in conjunction with an agreed, detailed operation procedure. It is essential that the real and imaginary parts of the complex refractive index of the material are indicated if the Mie theory is applied in data analysis. The representation of the particle-size distribution by volume will equal that of the distribution by mass, provided that the density of the particles is the same for all size fractions.
The response of a laser diffraction instrument meets the requirements if the mean value of x50 from at least three independent measurements does not deviate by more than 3% from the certified range of values of the certified reference material. The mean values for x10 and x90 must not deviate by more than 5% from the certified range of values. Below 10 µm, these values must be doubled.
Although the use of materials consisting of spherical particles is preferable, nonspherical particles may also be employed. Preferably, these particles have certified or typical values from laser diffraction analysis performed according to an agreed, detailed operating procedure. The use of reference values from methods other than laser diffraction may cause a significant bias. The reason for this bias is that the different principles inherent in the various methods may lead to different sphere-equivalent diameters for the same nonspherical particle.
Although the use of certified reference materials is preferred, other well-defined reference materials may also be employed. They consist of substances of typical composition and particle-size distribution for a specified class of substances. Their particle-size distribution has proven to be stable over time. The results must comply with previously determined data, with the same precision and bias as for the certified reference material.
Qualification of the System
In addition to the calibration, the performance of the instrument must be qualified at regular time intervals or as frequently as appropriate. This can be undertaken using any suitable reference material as mentioned in the previous paragraph.
The qualification of the system is based on the concept that the equipment, electronics, software, and analytical operations constitute an integral system, which can be evaluated as an entity. Thus the entire measurement procedure is examined, including sample collection, sample dispersion, sample transport through the measuring zone, and the measurement and deconvolution procedure. It is essential that the total operational procedure is fully described.
In general, unless otherwise specified in the individual monograph, the response of a laser diffraction instrument is considered to meet the requirements if the x50 value does not deviate by more than 10% from the range of values of the reference material. If optionally the values at the sides of the distribution are evaluated (e.g., x10 and x90), then these values must not deviate by more than 15% from the certified range of values. Below 10 µm, these values must be doubled.
Note—For calibration of the instrument, stricter requirements are laid down in the paragraph on Calibration.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Kahkashan Zaidi, Ph.D.
Senior Scientific Liaison 1-301-816-8269 |
(GCPA2010) General Chapters - Physical Analysis |
USP35–NF30 Page 176
Pharmacopeial Forum: Volume No. 35(3) Page 707