INTRODUCTION
Parametric release is defined as the release of terminally sterilized batches or lots of sterile products based upon compliance with the defined critical parameters of sterilization without having to perform the requirements under Sterility Tests  71
71 . Parametric release is a possibility when the mode of sterilization is very well understood, the physical parameters of processing are well defined, predictable, and measurable, and the lethality of the cycle has been microbiologically validated through the use of appropriate biological indicators or, in the case of ionizing radiation, the appropriate microbiological and dosimetric tests. The use of parametric release for sterilization processes requires prior FDA approval. It should be expected that the regulatory agencies evaluating submissions including the use of parametric product release would insist upon a well supported scientific rationale for the sterilization process and well documented validation data. The agencies would need assurance that any marketed sample of product will be sterile and if tested after release would pass the requirements for sterility as found in the general chapter Sterility Tests 71.
. Parametric release is a possibility when the mode of sterilization is very well understood, the physical parameters of processing are well defined, predictable, and measurable, and the lethality of the cycle has been microbiologically validated through the use of appropriate biological indicators or, in the case of ionizing radiation, the appropriate microbiological and dosimetric tests. The use of parametric release for sterilization processes requires prior FDA approval. It should be expected that the regulatory agencies evaluating submissions including the use of parametric product release would insist upon a well supported scientific rationale for the sterilization process and well documented validation data. The agencies would need assurance that any marketed sample of product will be sterile and if tested after release would pass the requirements for sterility as found in the general chapter Sterility Tests 71.
It is important to consider the limitations of the Sterility Tests 71 in the evaluation of terminally sterilized products. The sterility test described in general chapter 71 is limited in its sensitivity and is statistically ill-suited to the evaluation of terminally sterilized products given the exceedingly low probability of contaminated units. Therefore, once a sterilization process is fully validated and operates consistently, a combination of physical sterilization data such as accumulated lethality or dosimetry in combination with other methods, such as load monitors (e.g., biological indicators, thermochemical indicators, or physicochemical integrators), can provide more accurate information than the sterility test regarding the release of terminally sterilized product to the marketplace.
There are four modes of sterilization that theoretically and practically could qualify for parametric release: moist heat, dry heat, ethylene oxide, and ionizing radiation sterilization. This information chapter first will cover the general issues related to parametric release, regardless of the modes of sterilization, and then discuss some specific modes of sterilization. The chapter will not address the parametric release of terminally sterilized medical devices.
Terminally sterilized products represent the lowest risk category of sterile pharmaceutical products. Unlike products aseptically manufactured in a microbiologically controlled environment, terminally sterilized products are subjected to a sterilization process that imparts a measurable minimum sterility assurance level, or SAL. Because aseptic processing relies on exclusion of microbiological contamination and is not based upon lethality imparted on the product in its sealed container, it is not possible to estimate the SAL. It is important to note that in the case of aseptic processing, SAL can only be estimated from media fill contamination rates or other forms of risk assessment. In the case of terminal sterilization, it is possible to calculate a minimum SAL or Probability of Nonsterility (PNS) quite accurately. Therefore, the term SAL has different contextual meanings when used to describe aseptic rather than terminal processes, and it is important that this difference is fully understood by scientists and engineers working in the field of sterile product manufacturing and control. The terms PNS and SAL are often used interchangeably.
Terminally sterilized products must have a probability of nonsterility (PNS) of not more than one in a million units produced. This is often stated as a PNS or SAL of 10–6, or the probability of product bioburden surviving the sterilization process in any single unit of product is less than one in one million. The proof that a terminally sterilized product complies with the 10–6 PNS can be accomplished by several different sterilization cycle development approaches. The proper application of these methods requires extensive scientific knowledge regarding the sterilization method selected for use with a specific product.
The strategies used to validate a terminal sterilization process development fall into three categories:
- Bioburden-based process.
- Biological indicator/bioburden combined process.
- Overkill process.
The bioburden-based process requires extensive knowledge of product bioburden. It should be noted that several radiation dose-setting procedures involve establishing radiation processes on the basis of bioburden count and radiation resistance. This method requires that at least a 10–6 PNS be attained for bioburden by the sterilization process. This means that if the product bioburden action level is 10 microorganisms or one logarithm, at least seven logarithms of bioburden must be inactivated to assure a 10–6 PNS. The bioburden-based method requires the user to develop suitable critical control points within the process to control the bioburden titer. Products that readily permit bioburden survival require more controlled manufacturing environments and more precise in-process control. This process is better suited for cycle development for clean or ultra-clean products containing a consistently low level of colony forming units (cfu) per product unit with a low frequency of spore-forming microorganisms. Also, this process may be necessary to permit terminal sterilization of a product that may potentially lose key qualities or attributes as a result of a more rigorous sterilization process.
The microbiologist may find that formal hazard analysis procedures, such as Hazard Analysis Critical Control Point (HACCP), are useful in establishing appropriate manufacturing control conditions and in-process control parameters.
The biological indicator/bioburden combined process is generally used when the manufacturer desires a sterilization process that demonstrates the inactivation of high numbers of biological indicator microorganisms known to be resistant to the process. While the manufacturer may have preferred utilizing an overkill process, potential loss of some product attributes may occur in an overkill process thereby necessitating the use of a biological indicator/bioburden combined process. This process requires knowledge of the bioburden load on and in the product, and a database relative to the sterilization resistance of the bioburden. The relative resistance of the selected biological indicator to that of the bioburden must be established on or in the product. Frequently, biological indicators bearing approximately 106 spores with D121-value > 1 minute are used in the development of such processes. Fractional exposure cycles are generally conducted to determine the relative sterilization resistance (or D value) between product inoculated with the biological indicator microorganism(s) and frequently encountered bioburden. This process is frequently used for sterilization cycle development by manufacturers of terminally sterilized parenteral products and for ethylene oxide sterilization of medical devices.
The overkill process is frequently used when the article being sterilized is completely inert to the sterilizing agent and sterilization cycle conditions without any concern for loss of product attributes or quality. When using this process, some bioburden knowledge should be available to ensure that the materials are not adulterated before sterilization. These data may include product bioburden count data and knowledge concerning the prevalence of spore formers. The database for this process need not be as extensive as bioburden data required for the bioburden process or the biological indicator/bioburden process. Generally, process-resistant biological indicators containing approximately 106 spores are used to establish the effectiveness of the sterilization process. However, a spore population of N0 can be chosen to confirm adequate process lethality. Overkill is generally defined as a process that would deliver a minimum of F01 of 12 minutes (see Critical Operating Parameters below) and is demonstrated biologically based upon the spore log reduction of calibrated biological indicators.
GENERAL REVIEW
Validation of Sterilization Process
Parametric release first requires that the chosen sterilization process be designed and validated to achieve a 10–6 PNS. Validation of most sterilization processes includes the validation of physical parameters of the process and of its microbiological effectiveness through the use of biological indicators. However, the use of biological indicators for establishing or periodically validating gamma radiation sterilization processes is uncommon. Widely recognized biological indicator organisms are used in the validation of moist heat processes because they provide a means of comparing physically measured lethality data with biological lethality. There should be a reasonable correlation between physically measured lethality data (F0) and biological lethality as determined by the evaluation of the process with biological indicators.
The predictable effectiveness of bioburden-based terminal sterilization is based on the number and resistance of microorganisms on or in a product. For this reason, one component of parametric release is an active microbiology control program to monitor the count and sterilization resistance of product bioburden. Bioburden control and enumeration is of far less significance when the overkill process design is used. In many cases, overkill processes do not require extensive ongoing assessment of bioburden and require less in-process control of the manufacturing environment.
Sterilization Microbiology Control Program
The purpose of this control program is to ensure that the microbiological status of the product, prior to being terminally sterilized, has not significantly deviated from the established microbiological control level used for validation of the sterilization process. The microbiology control program includes the monitoring of the bioburden on or in the product and the monitoring of the microbiological status of any necessary containers, closures, or packaging materials. Also included is a program to evaluate the microbiological status of the environment where the product is processed. The control program is particularly important in cases where the terminal sterilization is not based on overkill, but rather on the bioburden or combined bioburden/biological indicator cycle development approach. In many cases, bioburden control and manufacturing environmental monitoring will not be required for overkill process designs, where the F0 of the process is at least 12 minutes. In other cases, even when overkill processes are employed, some limited monitoring will be needed. Monitoring of overkill processes for bioburden is generally limited to those products that support microbial growth. Of particular concern in this case is the potential for the product to be contaminated with microbial toxins or to be degraded by microorganisms.
The frequency of monitoring will depend on the variations of bioburden from potential sources. The number of microorganisms, their identification, as well as their resistance to the specified sterilization mode should be considered when parametric release of terminally sterilized product is established. Resistance to a specified sterilization mode by different species can influence sterilization effectiveness and the determination of sterilization process conditions when using the bioburden or combined bioburden/biological indicator method of cycle development. In the bioburden approach to process development, indicator organisms more resistant than typical bioburden may be used, although extreme differentials in resistance are not required. Information on the performance of biological indicators may be found in the general chapter Biological Indicators—Resistance Performance Tests 55.
Change Control System
Changes introduced to the sterilization processing equipment could result in a significant departure of the initially validated parametric release process. It is, therefore, essential that a change control system be instituted. A change control system is a formal system with appropriate standard operating procedures, which would include approval of changes in the sterilization processing equipment. This system would assess all the changes in relation to the critical parameters included in parametric release. The change control system also includes technical and management review and criteria for acceptance or revision of changes. If a change would significantly affect any critical parameter, each parameter would have to be revalidated in terms of sterility assurance of the pharmaceutical product to a minimum 10–6 PNS. Appropriate regulatory notification would also be part of the revalidation process.
Release Procedures
A quality assurance program should be established that describes in detail the batch or lot release steps for parametric release of sterilized products and the required documentation. Although the assessment of the sterility assurance of products is primarily based on measurement of physical process parameters, a number of areas should be reviewed, documented, and approved for the parametric release of these products. These areas may include the following: a review of batch records; a review of the ongoing microbiological environmental control program results and presterilization bioburden; and a review of records of thermographic data, load monitors, and results of critical and noncritical data that may have been used to demonstrate process control. It is also important to ensure that the sterilizer is current relative to calibration, maintenance, and revalidation.
The implementation and practice of parametric release is not an intermittent program. Once such a program is implemented, release of the sterilized product is made in accordance with the requirements of the regulatory approved program. Product release by other means is not acceptable if the predefined critical operational parameters are not achieved.
MODES OF STERILIZATION
Moist Heat Sterilization
Moist heat sterilization of pharmaceutical products includes several types of sterilizing environments and sterilizing media. Saturated steam, hot water spray, and submerged hot water processes are all considered as moist heat sterilizing environments. Different processes may be used to sterilize products by moist heat, and they include batch-type sterilizers and continuous-type sterilizers.
critical operating parameters
A defined list of key process parameters and their respective operating limits are defined and established in the sterilization process specifications. Critical operating parameters are those that are absolutely essential to ensure product sterilization to a 10–6 PNS. Examples of critical operating parameters may include, but are not limited to, dwell time limits, minimum and maximum limits for process peak dwell temperature, average peak dwell temperature, and the results of the batch or lot release test that satisfies the requirements of CFR, Part 211 (e.g., a load monitor results from the laboratory). F0 may be used as a critical parameter only when temperature and time relationships are well defined. Other measured parameters may be considered secondary (or noncritical) parameters and may include maximum and minimum time to peak dwell, chamber pressure, and if applicable, chamber water level, sterilizing water time above defined temperature limits, and recirculating water pump pressure differential.
Ethylene Oxide Sterilization
The application of parametric release of pharmaceutical products sterilized by ethylene oxide is more difficult than parametric release of products sterilized by moist heat processes. Critical parameters for ethylene oxide (ETO) sterilization are interrelated and more complex than moist heat processes.
critical operating parameters
Critical parameters may include the following: temperature, amount of relative humidity present, ethylene oxide concentration, overall exposure time, product and load density, and gas permeability factors.
Parametric release of pharmaceutical products can be achieved if an automated measurement system for the critical parameters is employed and sterilization loads are closely defined and validated relative to product types, densities, packaging materials, and overall load configurations. An example of the measurement of critical factors that may be considered for parametric release would be the use of calibrated ETO pressure recordings to provide an estimate of ETO concentration during the process hold time or the use of direct measurement of ETO concentration by IR or gas chromatography. Because of variances that might occur in the key parameters during sterilization, parametric release is not widely used for products sterilized by ETO.
However, to ensure parametric release, in addition to the attainment of process parameters of the ethylene oxide sterilization, biological indicators (and their sterility testing after sterilization processing) or the use of physicochemical integrators for the ethylene oxide sterilization are often used as load monitors (critical parameters).
Radiation Sterilization
Two radiation sterilizing processes have been used: gamma and electron beam sterilization (i.e., ionizing radiation). Some pharmaceutical products, either in bulk or in their finished formats, have been sterilized by radiation. In discussing the critical parameters of radiation sterilization necessary for parametric release, it is customary to refer to parametric release as dosimetric release. Dosimetric release is provided by the use of a chemical dosimeter that measures the delivery of a minimum specified radiation dosage, which has been shown to provide sterilization of the product to a minimum 10–6 PNS.
The use of a dosimeter in ionizing radiation sterilization measures delivery of a minimum absorbed radiation dose to a pre-established low dose zone in the irradiated product carrier. This will require mapping of the profile of absorbed ionizing radiation across the density ranges processed in the product carrier. The lowest specified radiation dosage for the process is correlated to predictable bioburden reduction levels by any one of the three documented methods.2 An alternative method may be considered whereby extensive product bioburden count and radiation resistance data are available. Dose verification studies would be conducted to ensure that the worst case bioburden load, relative to resistance and numbers, can be inactivated at the lowest dose zone in the carrier system to provide at least a 10–6 PNS. This method would of course require an ongoing program of bioburden assessment. The target for the radiation cycle is a minimum 10–6 PNS relative to the product bioburden. Dosimetric release of a radiation-sterilized product depends on the delivery of at least a minimum dosage; thus, the critical operational parameters that govern the delivery of that dosage must be within specified limits. These operational critical parameters may include the following: a stacking configuration within the radiation carrier, bulk density of the product, speed of the conveyor or carrier system, distance to the radiation source, duration of product exposure, and appropriate defined adjustments for a decaying radiation source. Demonstration of consistency in the absorbed radiation dosage at areas of minimum and maximum zones of radiation absorption within the fully loaded carriers on a batch-to-batch basis is a necessary condition for dosimetric release of radiation-sterilized pharmaceutical products.
SUMMARY
The conversion to parametric release in lieu of product sterility testing as described in general chapter Sterility Tests 71 requires prior FDA approval. Parametric release is advantageous for terminally sterilized products. The extensiveness of data required to establish parametric release, compared to the general chapter 71 procedures, which lack sensitivity to very low levels of microbial contamination, can result in a more accurate and reliable assessment of the probability of nonsterility of product lots.
1
F0 is defined as the calculated equivalent time (in minutes) of process lethality to time at 121.1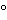 , assuming a Z value of 10.0 in the product being sterilized.
, assuming a Z value of 10.0 in the product being sterilized.
2
ANSI/AAMI/ISO 11137-1996, Sterilization of Health Care Products—Requirements for Validation and Routine Control—Radiation Sterilization, July 11, 1994.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Radhakrishna S Tirumalai, Ph.D.
Principal Scientific Liaison 1-301-816-8339 |
(GCM2010) General Chapters - Microbiology |
USP35–NF30 Page 870
Pharmacopeial Forum: Volume No. 30(5) Page 1741