Change to read:
Microbiologically controlled environments are used for a variety of purposes within the healthcare industry. This general information chapter provides information and recommendations for environments where the risk of microbial contamination is controlled through aseptic processing. Products manufactured in such environments include pharmaceutical sterile products, bulk sterile drug substances, sterile intermediates, excipients, and, in certain cases, medical devices. Aseptic processing environments are far more critical in terms of patient risk than controlled environments used for other manufacturing operations—for example, equipment and component preparation, limited bioburden control of nonsterile products, and processing of terminally sterilized products. In this chapter, the type of aseptic processing is differentiated by the presence or absence of human operators. An advanced aseptic process is one in which direct intervention with open product containers or exposed product contact surfaces by operators wearing conventional cleanroom garments is not required and never permitted. [Note—A description of terms used in this chapter can be found in the Appendix at the end of the chapter. ]
The guidance provided in this chapter and the monitoring parameters given for microbiological evaluation should be applied only to clean rooms, restricted-access barrier systems (RABS), and isolators used for aseptic processing. ISO-classified environments used for other purposes are not required to meet the levels of contamination control required for aseptically produced sterile products. The environments used for nonsterile applications require different microbial control strategies.
A large proportion of products labeled as sterile are manufactured by aseptic processing rather than terminal sterilization. Because aseptic processing relies on the exclusion of microorganisms from the process stream and the prevention of microorganisms from entering open containers during processing, product bioburden as well as the bioburden of the manufacturing environment are important factors governing the risk of unacceptable microbial contamination. The terms aseptic and sterile are not synonymous. Sterile means having a complete absence of viable microorganisms or organisms that have the potential to reproduce. In the purest microbiological sense, an aseptic process is one that prevents contamination by the exclusion of microorganisms. In contemporary aseptic healthcare-product manufacturing, aseptic describes the process for handling sterilized materials in a controlled environment designed to maintain microbial contamination at levels known to present minimal risk.
In any environment where human operators are present, microbial contamination at some level is inevitable. Even the most cautious clean-room environment design and operation will not eliminate the shedding of microorganisms if human operators are present. Thus, an expectation of zero contamination at all locations during every aseptic processing operation is technically not possible and thus is unrealistic. There are no means to demonstrate that an aseptic processing environment and the product-contact surfaces within that environment are sterile. Monitoring locations should be determined based upon a assessment of risk. Although manufacturers should review environmental monitoring results frequently to ensure that the facility operates in a validated state of control, monitoring results can neither prove nor disprove sterility. Because of the limitations of monitoring, manufacturers cannot rely directly on monitoring, statistics, or periodic aseptic-processing simulations to ensure a sterility assurance level.
Environmental monitoring is usually performed by personnel and thus requires operator intervention. As a result, environmental monitoring can both increase the risk of contamination and also give false-positive results. Thus, intensive monitoring is unwarranted, particularly in the ISO 5 environments that are used in the most critical zones of aseptic processing.
A number of sampling methods can be used to assess and control the microbiological status of controlled environments for aseptic processing. At present, nearly all of these methods rely on the growth and recovery of microorganisms, many of which can be in a damaged state caused by environmental stress and therefore may be difficult to recover. The numerical values for air, surface, and personnel monitoring included in this chapter are not intended to represent limits or specifications but are strictly informational. Because of the variety of microbiological sampling equipment and methods, it is not scientifically reasonable to suggest that the attainment of these values guarantees microbial control or that excursions beyond values in this chapter indicate a loss of control. The assessment of risks associated with manufacturing environments must be made over a significant period; and in each case, the contamination recovery rate metric should be established on the basis of a review of actual findings within the facility. The objective of each user should be to use contamination recovery rates to track ongoing performance and to refine the microbiological control program to foster improvements. When optimum operational conditions are achieved within a facility, contamination recovery rate levels typically become relatively stable within a normal range of variability.
There are no standard methods for air sampling, and available literature indicates that air-sampling methods are highly variable. It should not be assumed that similar sample volumes taken by different methods will produce similar rates of recovery. Many factors can affect microbial recovery and survival, and different air sampler suppliers may have designed their systems to meet different requirements. Also, sample-to-sample variation in microbial sampling can be extensive. Limited data are available regarding the accuracy, precision, sensitivity, and limits of detection of monitoring methods used in the aseptic processing of healthcare products.
Surface sampling methods are also not standardized. Different media are employed, and in the case of swabs, different results have been reported for wet and dry swab methods and contact plates. Replicate sample contact plates should be expected to give similar results under identical conditions, but rates of recovery have been reported to be both lower than expected and highly variable. In general, surface monitoring has been found to recover <50%, even when used with relatively high inoculum levels on standardized coupons. In actual production environments where organisms are stressed to varying degrees, recovery rates may be lower.
ADVANCED ASEPTIC TECHNOLOGIES
Advanced aseptic technologies can be defined as those that do not rely on the direct intervention of human operators during processing. At present, technologies such as isolators, blow/fill/seal, and closed RABS (designs that are never opened during setup or operation) may be considered advanced aseptic technologies, provided that direct intervention by gowned personnel is disallowed during processing. In recent years, isolator technology has found a broad acceptance in healthcare manufacturing. Isolators and closed RABS effectively separate the operator from the critical aseptic processing environment. Because these systems substantially reduce contamination risk, their microbiological control levels are higher than those of conventional clean rooms that have comparable particulate air classification level, for example, ISO 5.
CLEAN ROOM CLASSIFICATION FOR ASEPTIC PROCESSING ENVIRONMENTS
The design and construction of clean rooms and controlled environments are covered in ISO 14644 series. This standard defines the performance of a clean environment with respect to the concentration of total particulates per unit volume. ISO 14644-1 stipulates the total particulate counts allowed for a clean environment to meet the defined air quality classifications. The reader is referred to this standard regarding the design characteristics and certification of clean environments.
Pharmaceutical manufacturers are concerned with nonviable particulate contamination in injectable products (see Particulate Matter in Injections  788
788 ). Unlike microbial contamination in which experimental data suggest that humans are the only significant source, nonviable particulates can arise both from humans and from processing equipment. Studies indicate that gowned humans slough particulate and microbial contamination at a rather consistent rate. However, the relationship between microbial (viable) and nonviable contamination does not hold for particulates shed by processing equipment. Where equipment is the primary source of particulate matter, the resulting particulates are essentially all nonviable.
). Unlike microbial contamination in which experimental data suggest that humans are the only significant source, nonviable particulates can arise both from humans and from processing equipment. Studies indicate that gowned humans slough particulate and microbial contamination at a rather consistent rate. However, the relationship between microbial (viable) and nonviable contamination does not hold for particulates shed by processing equipment. Where equipment is the primary source of particulate matter, the resulting particulates are essentially all nonviable.
The argument that if fewer total particulates are present in a clean room, it is less likely that airborne microorganisms will be present is true only if human operators are the source of particulate matter. It is not possible to clearly distinguish between background total particulate contamination generated largely by mechanical operations and the total particulates contributed by personnel. Thus, it is both commonplace and proper for clean-room environmental monitoring programs to consist of both a total particulate component and a microbiological component. Table 1 describes the clean room classifications commonly used in the pharmaceutical industry. In aseptic processing, clean environments of ISO 14644-1 Classes 5–8 are typically used.
Table 1. Airborne Total Particulate Cleanliness Classesa
| ISO Classb | Particles |
|---|---|
| ISO 5 | 3520 |
| ISO 6 | 35,200 |
| ISO 7 | 352,000 |
| ISO 8 | 3,520,000 |
|
a
Taken from ISO International Standard 14644 Part 1, published by the International Organization for Standardization, May 1999.
b
The four ISO 14644-1 classes correspond closely to former U.S. Federal Standard 209E classifications. The relationships are
ISO 5/Class 100, ISO 6/Class 1000, ISO 7/Class 10,000, and ISO 8/Class 100,000. |
|
Isolators and closed RABS present a different picture, because personnel are excluded from the aseptic processing environment and manipulations are made using glove-and-sleeve assemblies and half-suits made of thick, flexible plastic (such as polyvinyl chloride or synthetic rubber). Personnel have far less effect on the microbial quality of the environment within an isolator enclosure than in clean room environments. Some users have chosen to operate RABS in a manner that allows open, direct human intervention. In an open operational state, these systems are more similar in operation to conventional clean rooms and therefore cannot be considered advanced aseptic processing systems. In an open RABS, the ability of operators to adversely affect microbial contamination risk is higher than with closed RABS or isolators.
Specifications for air changes per hour and air velocities are not included in ISO 14644, nor were they included in Federal Standard 209E. Typically, ISO Class 8/Class 100,000 rooms are designed to provide a minimum of 20 air changes per hour; ISO Class 7/Class 10,000 rooms are designed to provide more than 50 air changes per hour; and ISO Class 5/Class 100 clean rooms provide more than 100 air changes per hour. The design of some facility criteria may differ. By diluting and removing contaminants, large volumes of air are likely to reduce airborne contamination in aseptic production. Optimum conditions vary considerably, depending on process characteristics, particularly the amount of contamination derived from personnel. These specifications should be used only as a guide in the design and operation of clean rooms, because the precise correlations among air changes per hour, air velocity, and microbial control have not been satisfactorily established experimentally.
Manufacturers should maintain a predominantly unidirectional flow of air (either vertical or horizontal) in a staffed Class 5 clean room environment, particularly when products, product containers, and closures are exposed. In the evaluation of air movement within a clean room, studying airflow visually by smoke studies or other suitable means is probably more useful than using absolute measures of airflow velocity and change rates. Risk assessment models are another useful way of reducing contamination risk and should be considered.
Air velocity and change rates are far less important in isolators or closed RABS than in clean rooms because personnel are more carefully separated from the product, product containers, and closures. Air velocities substantially lower than those used in human-scale clean rooms have proved adequate in isolator systems and may be appropriate in RABS as well. In zones within isolators where particulate matter poses a hazard to product quality, predominantly vertical or horizontal unidirectional airflow can be maintained. Experience has shown that well-controlled mixing or turbulent airflow is satisfactory for many aseptic processes and for sterility testing within isolators (see Sterility Testing—Validation of Isolator Systems 1208).
IMPORTANCE OF A MICROBIOLOGICAL EVALUATION PROGRAM FOR CONTROLLED ENVIRONMENTS
Monitoring of total particulate count in controlled environments, even with the use of electronic instrumentation on a continuous basis, does not provide information on the microbiological content of the environment. The basic limitation of particulate counters is that they measure particles of 0.5 µm or larger. While airborne microorganisms are not free-floating or single cells, they frequently associate with particles of 10–20 µm. Particulate counts as well as microbial counts within controlled environments vary with the sampling location and the activities being conducted during sampling. Monitoring the environment for nonviable particulates and microorganisms is an important control function because they both are important in achieving product compendial requirements for Foreign and Particulate Matter and Sterility in Injections 1.
Total particulate monitoring may provide a better means of evaluating the overall quality of the environment in isolators and closed RABS than in most conventional clean rooms. The superior exclusion of human-borne contamination provided by an isolator results in an increased proportion of nonviable particulates. Total particulate counting in an isolator is likely to provide an immediate indicator of changes in contamination level. Microbial monitoring programs should assess the effectiveness of cleaning and sanitization practices by and of personnel who could have an impact on the bioburden. Because isolators are typically decontaminated using an automatic vapor or gas generation system, microbial monitoring is much less important in establishing their efficiency in eliminating bioburden. These automatic decontamination systems are validated directly, using an appropriate biological indicator challenge, and are controlled to defined exposure parameters during routine use to ensure consistent decontamination.
Microbial monitoring cannot and need not identify and quantify all microbial contaminants in these controlled environments. Microbiological monitoring of a clean room is technically a semiquantitative exercise, because a truly quantitative evaluation of the environment is not possible, given the limitations in sampling equipment. Both the lack of precision of enumeration methods and the restricted sample volumes that can be effectively analyzed suggest that environmental monitoring is incapable of providing direct quantitative information about sterility assurance. Analysts should remember that no microbiological sampling plan can prove the absence of microbial contamination, even when no viable contamination is recovered. The absence of growth on a microbiological sample means only that growth was not discovered; it does not mean that the environment is free of contamination.
Routine microbial monitoring should provide sufficient information to demonstrate that the aseptic processing environment is operating in an adequate state of control. The real value of a microbiological monitoring program lies in its ability to confirm consistent, high-quality environmental conditions at all times. Monitoring programs can detect changes in the contamination recovery rate that may be indicative of changes in the state of control within the environment.
Environmental microbial monitoring and analysis of data by qualified personnel can assist in ensuring that a suitable state of control is maintained. The environment should be sampled during normal operations to allow the collection of meaningful, process-related data. Microbial sampling should occur when materials are in the area, processing activities are ongoing, and a full complement of personnel is working within the aseptic processing environment.
Microbial monitoring of manufacturing clean rooms, RABS, and isolators should include compressed gases, surfaces, room or enclosure air, and any other materials and equipment that might produce a risk of contamination. The analysis of contamination trends in an aseptic environment has long been a component of the environmental control program. In aseptic processing environments and particularly in ISO Class 5 environments, contamination is infrequently observed. In isolator enclosures, contamination is rarer still because of superior exclusion of human-borne contamination. Because of the criticality of these environments, even minor changes in the contamination incident rates may be significant, and manufacturers should frequently and carefully review monitoring data. In less critical environments, microbial contamination may be higher, but changes in recovery rates should be noted, investigated, and corrected. Isolated recoveries of microorganisms should be considered a normal phenomenon in conventional clean rooms, and these incidents generally do not require specific corrective action, because it is almost certain that investigations will fail to yield a scientifically verifiable cause. Because sampling itself requires an aseptic intervention in conventional clean rooms, any single uncorrelated contamination event could be a false positive.
When contamination recovery rates increase from an established norm, process and operational investigation should take place. Investigations will differ depending on the type and processing of the product manufactured in the clean room, RABS, or isolator. Investigation should include a review of area maintenance documentation; sanitization/decontamination documentation; the occurrence of nonroutine events; the inherent physical or operational parameters, such as changes in environmental temperature and relative humidity; and the training status of personnel.
In closed RABS and isolator systems, the loss of glove integrity or the accidental introduction of material that has not been decontaminated are among the most probable causes of detectable microbial contamination. Following the investigation, actions should be taken to correct or eliminate the most probable causes of contamination. Because of the relative rarity of contamination events in modern facilities, the investigation often proves inconclusive. When corrective actions are undertaken, they may include reinforcement of personnel training to emphasize acceptable gowning and aseptic techniques and microbial control of the environment. Some additional microbiological sampling at an increased frequency may be implemented, but this may not be appropriate during aseptic processing because intrusive or overly intensive sampling may entail an increased contamination risk. When additional monitoring is desirable, it may be more appropriate during process simulation studies. Other measures that can be considered to better control microbial contamination include additional sanitization, use of different sanitizing agents, and identification of the microbial contaminant and its possible source.
In any aseptic environment, conventional or advanced, the investigation and the rationale for the course of action chosen as a result of the investigation must be carefully and comprehensively documented.
PHYSICAL EVALUATION OF CONTAMINATION CONTROL EFFECTIVENESS
Clean environments should be certified as described in ISO 14644 series in order to meet their design classification requirements. The design, construction, and operation of clean rooms vary greatly, so it is difficult to generalize requirements for parameters such as filter integrity, air velocity, air patterns, air changes, and pressure differential. In particularly critical applications such as aseptic processing, a structured approach to physical risk assessment may be appropriate.
One such method has been developed by Ljundqvist and Reinmüller. This method, known as the L-R method, challenges the air ventilation system by evaluating both airflow and the ability of an environment to dilute and remove airborne particles. In the L-R method, a smoke generator allows analysts to visualize the air movements throughout a clean room or a controlled environment, including vortices or turbulent zones, and the airflow pattern can be fine-tuned to minimize these undesirable effects. Following visual optimization of airflow, particulate matter is generated close to the critical zone and sterile field. This evaluation is done under simulated production conditions but with equipment and personnel in place. This type of test can also be used to evaluate the ability of RABS and isolator systems, particularly around product exit ports in these systems, to resist the effects of contamination.
Visual evaluation of air movement within clean rooms is a subjective process. Complete elimination of turbulence or vortices is not possible in operationing clean rooms that contain personnel and equipment. Air visualization is simply one step in the effort to optimize clean room operations and is not a definitive pass/fail test, because acceptable or unacceptable conditions are not readily definable.
Proper testing and optimization of the physical characteristics of the clean room, RABS, or isolator are essential before implementation of the microbiological monitoring program. Assurance that the clean room, RABS, or isolator is in compliance with its predetermined engineering specifications provides confidence that the ability of the facility systems and operating practices to control the bioburden and nonviable particulate matter are appropriate for the intended use. These tests should be repeated during routine certification of the clean room or advanced aseptic processing systems, and whenever significant changes are made to the operation, such as personnel flow, equipment operation, material flow, air-handling systems, or equipment layout.
TRAINING OF PERSONNEL
Good personnel performance plays an essential role in the control of contamination, proper training and supervision are central to contamination control. Aseptic processing is the most critical activity conducted in microbiological controlled environments, and manufacturers must pay close attention to details in all aspects of this endeavor. Rigorous discipline and strict supervision of personnel are essential in order to ensure a level of environmental quality appropriate for aseptic processing.
Training of all personnel working in controlled environments is critical. This training is equally important for personnel responsible for the microbial monitoring program, because contamination of the clean working area could inadvertently occur during microbial sampling. In highly automated operations, monitoring personnel may be the employees who have the most direct contact with the critical surfaces and zones within the processing area. Microbiological sampling has the potential to contribute to microbial contamination caused by inappropriate sampling techniques or by placing personnel in or near the critical zone. A formal training program is required to minimize this risk. This training should be documented for all personnel who enter controlled environments. Interventions should always be minimized, including those required for monitoring activities; but when interventions cannot be avoided, they must be conducted with aseptic technique that approaches perfection as closely as possible.
Management of the facility must ensure that personnel involved in operations in clean rooms and advanced aseptic processing environments are well versed in relevant microbiological principles. The training should include instruction about the basic principles of aseptic technique and should emphasize the relationship of manufacturing and handling procedures to potential sources of product contamination. Those supervising, auditing, or inspecting microbiological control and monitoring activities should be knowledgeable about the basic principles of microbiology, microbial physiology, disinfection and sanitation, media selection and preparation, taxonomy, and sterilization. The staff responsible for supervision and testing should have academic training in medical or environmental microbiology. Sampling personnel as well as individuals working in clean rooms should be knowledgeable about their responsibilities in minimizing the release of microbial contamination. Personnel involved in microbial identification require specialized training about required laboratory methods. Additional training about the management of collected data must be provided. Knowledge and understanding of applicable standard operating procedures are critical, especially those procedures relating to corrective measures taken when environmental conditions require. Understanding of contamination control principles and each individual's responsibilities with respect to good manufacturing practices (GMPs) should be an integral part of the training program, along with training in conducting investigations and in analyzing data.
The only significant sources of microbial contamination in aseptic environments are the personnel. Because operators disperse contamination and because the ultimate objective in aseptic processing is to reduce end-user risk, only healthy individuals should be permitted access to controlled environments. Individuals who are ill must not be allowed to enter an aseptic processing environment, even one that employs advanced aseptic technologies such as isolators, blow/fill/seal, or closed RABS.
The importance of good personal hygiene and a careful attention to detail in aseptic gowning cannot be overemphasized. Gowning requirements differ depending on the use of the controlled environment and the specifics of the gowning system itself. Aseptic processing environments require the use of sterilized gowns with the best available filtration properties. The fullest possible skin coverage is desirable, and sleeve covers or tape should be considered to minimize leaks at the critical glove–sleeve junction. Exposed skin should never be visible in conventional clean rooms under any conditions. The personnel and gowning considerations for RABS are essentially identical to those for conventional clean rooms.
Once employees are properly gowned, they must be careful to maintain the integrity of their gloves, masks, and other gown materials at all times. Operators who work with isolator systems are not required to wear sterilized clean-room gowns, but inadequate aseptic technique and employee-borne contamination are the principal hazards to safe aseptic operations in isolators, as well as RABS, and in conventional clean rooms. Glove-and-sleeve assemblies can develop leaks that can allow the mechanical transfer of microorganisms to the product. A second glove, worn either under or over the primary isolator/RABS glove, can provide an additional level of safety against glove leaks or can act as a hygienic measure. Also, operators must understand that aseptic technique is an absolute requirement for all manipulations performed with gloves within RABS and isolator systems.
The environmental monitoring program, by itself, cannot detect all events in aseptic processing that might compromise the microbiological quality of the environment. Therefore, periodic media-fill or process simulation studies are necessary, as is thorough ongoing supervision, to ensure that appropriate operating controls and training are effectively maintained.
CRITICAL FACTORS IN THE DESIGN AND IMPLEMENTATION OF A MICROBIOLOGICAL ENVIRONMENTAL MONITORING PROGRAM
Since the advent of comprehensive environmental monitoring programs, their applications in capturing adverse trends or drifts has been emphasized. In a modern aseptic processing environment—whether an isolator, RABS, or conventional clean room—contamination has become increasingly rare. Nevertheless, a monitoring program should be able to detect a change from the validated state of control in a facility and to provide information for implementing appropriate countermeasures. An environmental monitoring program should be tailored to specific facilities and conditions. It is also helpful to take a broad perspective in the interpretation of data. A single uncorrelated result on a given day may not be significant in the context of the technical limitations associated with aseptic sampling methods.
Selection of Growth Media
A general microbiological growth medium such as soybean–casein digest medium (SCDM) is suitable for environmental monitoring in most cases because it supports the growth of a wide range of bacteria, yeast, and molds. This medium can be supplemented with additives to overcome or to minimize the effects of sanitizing agents or of antibiotics. Manufacturers should consider the specific detection of yeasts and molds. If necessary, general mycological media such as Sabouraud’s, modified Sabouraud’s, or inhibitory mold agar can be used. In general, monitoring for strict anaerobes is not performed, because these organisms are unlikely to survive in ambient air. However, micro-aerophilic organisms may be observed in aseptic processing. Should anoxic conditions exist or if investigations warrant (e.g., identification of these organisms in sterility testing facilities or Sterility Tests 71 results), monitoring for micro-aerophiles and organisms that grow under low-oxygen conditions may be warranted. The ability of any media used in environmental monitoring, including those selected to recover specific types of organisms, must be evaluated for their ability to support growth, as indicated in 71.
Selection of Culture Conditions
Time and incubation temperatures are set once the appropriate media have been selected. Typically, for general microbiological growth media such as SCDM, incubation temperatures in the ranges of approximately 20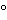 –35 have been used with an incubation time of not less than 72 hours. Longer incubation times may be considered when contaminants are known to be slow growing. The temperature ranges given above are by no means absolute. Mesophilic bacteria and mold common to the typical facility environment are generally capable of growing over a wide range of temperatures. For many mesophilic organisms, recovery is possible over a range of approximately 20. In the absence of confirmatory evidence, microbiologists may incubate a single plate at both a low and a higher temperature. Incubating at the lower temperature first may compromise the recovery of Gram-positive cocci that are important because they are often associated with humans.
–35 have been used with an incubation time of not less than 72 hours. Longer incubation times may be considered when contaminants are known to be slow growing. The temperature ranges given above are by no means absolute. Mesophilic bacteria and mold common to the typical facility environment are generally capable of growing over a wide range of temperatures. For many mesophilic organisms, recovery is possible over a range of approximately 20. In the absence of confirmatory evidence, microbiologists may incubate a single plate at both a low and a higher temperature. Incubating at the lower temperature first may compromise the recovery of Gram-positive cocci that are important because they are often associated with humans.
Sterilization processes for preparing growth media should be validated. When selective media are used for monitoring, incubation conditions should reflect published technical requirements. Contamination should not be introduced into a manufacturing clean room as a result of using contaminated sampling media or equipment. Of particular concern is the use of aseptically prepared sampling media. Wherever possible, sampling media and their wrappings should be terminally sterilized by moist heat, radiation, or other suitable means. If aseptically prepared media must be used, analysts must carry out preincubation and visual inspection of all sampling media before introduction into the clean room. The reader is referred to Microbiological Best Laboratory Practices 1117 for further information regarding microbiology laboratory operations and control.
ESTABLISHMENT OF SAMPLING PLAN AND SITES
During initial startup or commissioning of a clean room or other controlled environment, specific locations for air and surface sampling should be determined. Locations considered should include those in proximity of the exposed product, containers, closures, and product contact surfaces. In aseptic processing, the area in which containers, closures, and product are exposed to the environment is often called the critical zone—the critical zone is always ISO 5. For aseptic operations the entire critical zone should be treated as a sterile field. A nonsterile object, including the gloved hands of clean room personnel or an RABS/isolator glove, should never be brought into contact with a sterile product, container closure, filling station, or conveying equipment before or during aseptic processing operations. Operators and environmental monitoring personnel should never touch sterile parts of conveyors, filling needles, parts hoppers, or any other equipment that is in the product-delivery pathway. This means that surface monitoring on these surfaces is best done at the end of production operations.
The frequency of sampling depends on the manufacturing process conducted within an environment. Classified environments that are used only to provide a lower overall level of bioburden in nonsterile product manufacturing areas require relatively infrequent environmental monitoring. Classified environments in which closed manufacturing operations are conducted, including fermentation, sterile API processing, and chemical processes, require fewer monitoring sites and less frequent monitoring because the risk of microbial contamination from the surrounding environment is comparatively low. Microbiological monitoring of environments in which products are filled before terminal sterilization is generally less critical than the monitoring of aseptic processing areas. The amount of monitoring required in filling operations for terminal sterilization depends on the susceptibility of the product survival and the potential for proliferation of microbial contamination. The identification and estimated number of microorganisms that are resistant to the subsequent sterilization may be more critical than the microbiological monitoring of the surrounding manufacturing environments.
It is not possible to recommend microbial control levels for each type of manufacturing environment. The levels established for one ISO Class 7 environment, for example, may be inappropriate for another ISO Class 7 environment, depending on the production activities undertaken in each. The user should conduct a prospective risk analysis and develop a rationale for the sampling locations and frequencies for each controlled environment. The classification of a clean room helps establish control levels, but that does not imply that all rooms of the same classification should have the same control levels and the same frequency of monitoring. Monitoring should reflect the microbiological control requirements of manufacturing or processing activities. Formal risk assessment techniques can result in a scientifically valid contamination control program.
Table 2 suggests frequencies of sampling in decreasing order of frequency and in relation to the criticality or product risk of the area being sampled. This table distinguishes between aseptic processing where personnel are aseptically gowned and those where a lesser gowning is appropriate. Environmental monitoring sampling plans should be flexible with respect to monitoring frequencies, and sample plan locations should be adjusted on the basis of the observed rate of contamination and ongoing risk analysis. On the basis of long-term observations, manufacturers may increase or decrease sampling at a given location or eliminate a sampling location altogether. Oversampling can be as deleterious to contamination control as undersampling, and careful consideration of risk and reduction of contamination sources can guide the sampling intensity.
Table 2. Suggested Frequency of Sampling for Aseptic
Processing Areasa
Processing Areasa
| Sampling Area/Location | Frequency of Sampling |
|---|---|
| Clean Room/RABS | |
| Critical zone (ISO 5 or better) | |
| Active air sampling | Each operational shift |
| Surface monitoring | At the end of the operation |
| Aseptic area adjacent critical zone | |
| All sampling | Each operating shift |
| Other nonadjacent aseptic areas | |
| All sampling | Once per day |
| Isolators | |
| Critical zone (ISO 5 or better) | |
| Active air sampling | Once per day |
| Surface monitoring | At the end of the campaign |
| Nonaseptic areas surrounding the isolator | |
| All sampling | Once per month |
|
a
All operators are aseptically gowned in these environments (with the exception of background environments for isolators). These recommendations do not apply to production areas for nonsterile products or other classified environments in which fully aseptic gowns are not donned.
|
|
SELECTION OF SAMPLE SITES WITHIN CLEAN ROOMS AND ASEPTIC PROCESSING AREAS
ISO 14644 suggests a grid approach for the total particulate air classification of clean rooms. This approach is appropriate for certifying the total particulate air quality performance against its design objective. Grids may also have value in analyzing risk from microbial contamination, although in general, grids that have no personnel activity are likely to have low risk of contamination. Microbial contamination is strongly associated with personnel, so microbiological monitoring of unstaffed environments is of limited value.
Microbiological sampling sites are best selected with consideration of human activity during manufacturing operations. Careful observation and mapping of the clean room during the qualification phase can provide useful information concerning the movement and positioning of personnel. Such observation can also yield important information about the most frequently conducted manipulations and interventions.
The location and movement of personnel within the clean room correlate with contamination risk to the environment and to the processes conducted within that environment. Sample sites should be selected so that they evaluate the impact of personnel movement and work within the area, particularly interventions and manipulations within the critical zone.
The most likely route of contamination is airborne, so the samples most critical to risk assessment are those that relate to airborne contamination near exposed sterile materials. Other areas of concern are entry points where equipment and materials move from areas of lower classification to those of higher classification. Areas within and around doors and airlocks should be included in the monitoring scheme. It is customary to sample walls and floors, and indeed sampling at these locations can provide information about the effectiveness of the sanitization program. Sampling at these locations can take place relatively infrequently, because contamination there is unlikely to affect product. Operators should never touch floors and walls, so mechanical transmission of contamination from these surfaces to critical areas where product is exposed should not occur.
Manufacturers typically monitor surfaces within the critical zone, although this should be done only at the end of operations. Residues of media or diluent from wet swabs should be avoided on surfaces, because they could lead to microbial proliferation. Also, cleaning surfaces to remove diluent or media requires personnel intervention and movements that can result in release of microbial contamination into the critical zone and can disrupt airflow.
MICROBIOLOGICAL CONTROL PARAMETERS IN CLEAN ROOMS, ISOLATORS, AND RABS
Since the early 1980s, manufacturers have established alert and action levels for environmental monitoring. In recent years the numerical difference between alert and action levels has become quite small, especially in ISO 5 environments. Growth and recovery in microbiological assays have normal variability in the range of ±0.5 log10. Studies on active microbiological air samplers indicate that variability of as high as tenfold is possible among commonly used sampling devices. As a result of this inherent variability and indeterminate sampling error, the supposed differences between, for example, an alert level of 1 cfu and an action level of 3 cfu are not analytically significant. Treating differences that are within expected, and therefore, normal ranges as numerically different is not scientifically valid and can result in unwarranted activities. In a practical sense, numerical values that vary by as much as five- to tenfold may not be significantly different.
Because of the limited accuracy and precision of microbial growth and recovery assays, analysts can consider the frequency with which contamination is detected rather than absolute numbers of cfu detected in any single sample. Also, a cfu is not a direct enumeration of microorganisms present but rather is a measure of contamination that may have originated from a clump of organisms.
Mean contamination recovery rates should be determined for each clean room environment, and changes in contamination recovery rate at a given site or within a given room may indicate the need for corrective action. Within the ISO 5 critical zone, airborne and surface contamination recovery rates of 1% or less should be attainable with current methods. Contamination recovery rates for closed RABS and isolator systems should be significantly lower still and can be expected to be <0.1%, on the basis of published monitoring results.
Contamination observed at multiple sites in an environment within a single sampling period may indicate increased risk to product and should be carefully evaluated. The appearance of contamination nearly simultaneously at multiple sites could also arise from poor sampling technique, so careful review is in order before drawing conclusions about potential loss of control. Resampling an environment several days after contamination is of little value, because the conditions during one sampling occasion may not be accurately duplicated during another.
Surface samples may also be taken from clean room garments. Personnel sampling should be emphasized during validation and is best done at the completion of production work in order to avoid adventitious contamination of the garments. In this case the average should be <1% for these sample sites as well. Gloves on closed RABS and isolators should meet the more rigorous expectation of <0.1% contamination recovery rates.
Because of the inherent variability of microbial sampling methods, contamination recovery rates are a more useful measure of trending results than is focusing on the number of colonies recovered from a given sample. Table 3 provides recommended contamination recovery rates for aseptic processing environments. The incident rate is the rate at which environmental samples are found to contain microbial contamination. For example, an incident rate of 1% would mean that only 1% of the samples taken have any contamination regardless of colony number. In other words, 99% of the samples taken are completely free of contamination. Contamination recovery rates that are higher than those recommended in Table 3 may be acceptable in rooms of similar classification that are used for lower-risk activities. Action should be required when the contamination recovery rate trends above these recommendations for a significant time.
Table 3. Suggested Initial Contamination Recovery Rates in Aseptic Environmentsa
| Room Classification |
Active Air Sample (%) |
Settle Plate (9 cm) 4 h Exposure (%) | Contact Plate or Swab (%) | Glove or Garment (%) |
|---|---|---|---|---|
| Isolator/Closed RABS (ISO 5 or better) |
<0.1 | <0.1 | <0.1 | <0.1 |
| ISO 5 | <1 | <1 | <1 | <1 |
| ISO 6 | <3 | <3 | <3 | <3 |
| ISO 7 | <5 | <5 | <5 | <5 |
| ISO 8 | <10 | <10 | <10 | <10 |
|
a
All operators are aseptically gowned in these environments (with the exception of background environments for isolators). These recommendations do not apply to production areas for nonsterile products or other classified environments in which fully aseptic gowns are not donned.
|
||||
Detection frequency should be based on actual monitoring data and should be retabulated monthly. Action levels should be based on empirical process capability. If detection frequencies exceed the recommendations in Table 3 or are greater than established process capability, then corrective actions should be taken. Corrective actions may include but are not limited to the following:
- Revision of the sanitization program, including selection of antimicrobial agents, application methods, and frequencies
- Increased surveillance of personnel practices, possibly including written critiques of aseptic methods and techniques
- Review of microbiological sampling methods and techniques
When higher-than-typical recovery levels for glove and garment contamination are observed, additional training for gowning practices may be indicated.
SIGNIFICANT EXCURSIONS
Excursions beyond approximately 15 cfu recovered from a single ISO 5 sample, whether from airborne, surface, or personnel sources, should happen very infrequently. When such ISO 5 excursions do occur, they may be indicative of a significant loss of control when they occur within the ISO 5 critical zone in close proximity to product and components. Thus, any ISO 5 excursion >15 cfu should prompt a careful and thorough investigation.
A key consideration for an abnormally high number of recovered colonies is whether this incident is isolated or can be correlated with other recoveries. Microbiologists should review recovery rates for at least two weeks before the incident of abnormally high recovery so that they can be aware of other recoveries that might indicate an unusual pattern. Microbiologists should carefully consider all recoveries, including those that are in the more typical range of 1–5 cfu. The identity of the organisms recovered is an important factor in the conduct of this investigation.
In the case of an isolated single excursion, establishing a definitive cause probably will not be possible, and only general corrective measures can be considered. It is never wise to suggest a root cause for which there is no solid scientific evidence. Also, there should be an awareness of the variability of microbial analysis. Realistically, there is no scientific reason to treat a recovery of 25 cfu as statistically different from a recovery of 15 cfu. A value of 15 cfu should not be considered significant in terms of process control, because realistically there is no difference between a recovery of 14 cfu and one of 15 cfu. Microbiologists should use practical scientific judgment in their approach to excursions.
FURTHER CONSIDERATIONS ABOUT DATA INTERPRETATION
In the high-quality environments required for aseptic processing, detection frequency typically is low. As can be seen from the rates recommended in Table 3, the majority of samples taken in an aseptic processing area will yield a recovery of zero contamination. In the most critical areas within an aseptic processing operation, it is expected that less than 1% of the samples will yield any recoverable contamination. In the most advanced of modern aseptic operations that use separative technologies such as isolators or closed RABS, the recovery rate will approach zero at all times.
The microbiologist responsible for environmental control or sterility assurance should not take this to mean that the environmental quality approaches sterility. The sensitivity of any microbial sampling system in absolute terms is not known. In environmental monitoring, a result of zero means only that the result is below the limit of detection of the analytical system. A false sense of security should not be derived from the infrequency of contamination recovery in aseptic processing.
Sterility assurance is best accomplished by a focus on human-borne contamination and the facility design features that best mitigate risk from this contamination. Greatest risk mitigation can be attained by reducing or eliminating human interventions through proper equipment design and by providing sufficient air exchanges per hour for the intended personnel population of the facility. Other risk mitigation factors include effective personnel and material movement and the proper control of temperature and humidity. Secondary factors for risk mitigation include cleaning and sanitization. Risk analysis models that analyze processes prospectively to reduce human-borne contamination risk by minimizing operator interventions are more powerful tools for sterility assurance than monitoring. Environmental monitoring cannot prove or disprove in absolute terms the sterility of a lot of product. Environmental monitoring can only assure those responsible for a process that a production system is in a consistent, validated state of control. Care should be taken to avoid drawing inappropriate conclusions from monitoring results.
SAMPLING AIRBORNE MICROORGANISMS
Among the most commonly used tools for monitoring aseptic environments are impaction and centrifugal samplers. A number of commercially available samplers are listed for informational purposes. The selection, appropriateness, and adequacy of using any particular sampler are the responsibility of the user.
Slit-to-Agar Air Sampler (STA):
The unit is powered by an attached source of controllable vacuum. The air intake is obtained through a standardized slit below which is placed a slowly revolving Petri dish that contains a nutrient agar. Airborne particles that have sufficient mass impact the agar surface, and viable organisms are allowed to grow. A remote air intake is often used to minimize disturbance of unidirectional airflow.
Sieve Impactor:
This apparatus consists of a container designed to accommodate a Petri dish that contains a nutrient agar. The cover of the unit is perforated with openings of a predetermined size. A vacuum pump draws a known volume of air through the cover, and airborne particles that contain microorganisms impact the agar medium in the Petri dish. Some samplers feature a cascaded series of sieves that contain perforations of decreasing size. These units allow determination of the size range distribution of particulates that contain viable microorganisms based on the size of the perforations through which the particles landed on the agar plates.
Centrifugal Sampler:
The unit consists of a propeller or turbine that pulls a known volume of air into the unit and then propels the air outward to impact on a tangentially placed nutrient agar strip set on a flexible plastic base.
Sterilizable Microbiological Atrium:
The unit is a variant of the single-stage sieve impactor. The unit's cover contains uniformly spaced orifices approximately 0.25 inch in size. The base of the unit accommodates one Petri dish containing a nutrient agar. A vacuum pump controls the movement of air through the unit, and a multiple-unit control center as well as a remote sampling probe are available.
Surface Air System Sampler:
This integrated unit consists of an entry section that accommodates an agar contact plate. Immediately behind the contact plate is a motor and turbine that pulls air through the unit's perforated cover over the agar contact plate and beyond the motor, where it is exhausted. Multiple mounted assemblies are also available.
Gelatin Filter Sampler:
The unit consists of a vacuum pump with an extension hose terminating in a filter holder that can be located remotely in the critical space. The filter consists of random fibers of gelatin capable of retaining airborne microorganisms. After a specified exposure time, the filter is aseptically removed and dissolved in an appropriate diluent and then plated on an appropriate agar medium to estimate its microbial content.
Settling Plates:
This method is still widely used as a simple and inexpensive way to qualitatively assess the environments over prolonged exposure times. Published data indicate that settling plates, when exposed for 4- to 5-hour periods, can provide a limit of detection for a suitable evaluation of the aseptic environment. Settling plates may be particularly useful in critical areas where active sampling could be intrusive and a hazard to the aseptic operation.
One of the major drawbacks of mechanical air samplers is the limited sample size of air being tested. When the microbial level in the air of a controlled environment is expected to contain extremely low levels of contamination per unit volume, at least 1 cubic meter of air should be tested in order to maximize sensitivity. Typically, slit-to-agar devices have an 80-L/min sampling capacity (the capacity of the surface air system is somewhat higher). If 1 cubic meter of air were tested, then it would require an exposure time of 15 min. It may be necessary to use sampling times in excess of 15 min to obtain a representative environmental sample. Although some samplers are reported to have high sampling volumes, consideration should be given to the potential for disruption of the airflow patterns in any critical area and to the creation of turbulence.
Technicians may wish to use remote sampling systems in order to minimize potential risks resulting from intervention by environmental samplers in critical zones. Regardless of the type of sampler used, analysts must determine that the extra tubing needed for a remote probe does not reduce the method's sensitivity to such an extent that detection of low levels of contamination becomes unlikely or even impossible.
SURFACE SAMPLING
Another component of the microbial-control program in controlled environments is surface sampling of equipment, facilities, and personnel. The standardization of surface sampling methods and procedures has not been as widely addressed in the pharmaceutical industry as has the standardization of air-sampling procedures. Surface sampling can be accomplished by the use of contact plates or by the swabbing method.
Contact plates filled with nutrient agar are used for sampling regular or flat surfaces and are directly incubated for the appropriate time and temperature for recovery of viable organisms. Specialized agar can be used for the recovery of organisms that have specific growth requirements. Microbial estimates are reported per contact plate.
The swabbing method can be used to supplement contact plates for sampling of irregular surfaces, especially irregular surfaces of equipment. The area that will be swabbed is defined with a sterile template of appropriate size. In general, it is in the range of 24–30 cm2. After sample collection the swab is placed in an appropriate diluent or transport medium and is plated onto the desired nutrient agar. The microbial estimates are reported per swab of defined sampling area.
Surface monitoring is used as an environmental assessment tool in all types of classified environments. In ISO 5 environments for aseptic processing, surface monitoring is generally performed beside critical areas and surfaces. Component hoppers and feed chutes that contact sterile surfaces on closures and filling needles can be tested for microbial contamination. Often in conventional staffed clean rooms, these product contact surfaces are steam sterilized and aseptically assembled. The ability of operators to perform these aseptic manipulations are evaluated during process stimulations or media fills, although true validation of operator technique in this manner is not possible. Surface monitoring on surfaces that directly contact sterile parts or product should be done only after production operations are completed. Surface sampling is not a sterility test and should not be a criterion for the release or rejection of product. Because these samples must be taken aseptically by personnel, it is difficult to establish with certainty that any contamination recovered is product related.
CULTURE MEDIA AND DILUENTS
The type of medium, liquid or solid, used for sampling or plating microorganisms depends on the procedure and equipment used. Any medium used should be evaluated for suitability for the intended purpose. The most commonly used all-purpose solid microbiological growth medium is soybean–casein digest agar. As previously noted, this medium can be supplemented with chemicals that counteract the effect of various antimicrobials.
IDENTIFICATION OF MICROBIAL ISOLATES
A successful environmental control program includes an appropriate level of identification of the flora obtained by sampling. A knowledge of the flora in controlled environments aids in determining the usual microbial flora anticipated for the facility and in evaluating the effectiveness of the cleaning and sanitization procedures, methods, agents, and recovery methods. The information gathered by an identification program can be useful in the investigation of the source of contamination, especially when recommended detection frequencies are exceeded.
Identification of isolates from critical and immediately adjacent areas should take precedence over identification of microorganisms from noncritical areas. Identification methods should be verified, and ready-to-use kits should be qualified for their intended purpose.
CONCLUSION
Environmental monitoring is one of several key elements required in order to ensure that an aseptic processing area is maintained in an adequate level of control. Monitoring is a qualitative exercise, and even in the most critical applications such as aseptic processing, conclusions regarding lot acceptability should not be made on the basis of environmental sampling results alone. Environments that are essentially free of human operators generally have low initial contamination rates and maintain low levels of microbial contamination. Human-scale clean rooms present a very different picture. Studies conclusively show that operators, even when carefully and correctly gowned, continuously slough microorganisms into the environment. Therefore, it is unreasonable to assume that samples producing no colonies, even in the critical zone or on critical surfaces, will always be observed. Periodic excursions are a fact of life in human-scale clean rooms, but the contamination recovery rate, particularly in ISO 5 environments used for aseptic processing, should be consistently low.
Clean-room operators, particularly those engaged in aseptic processing, must strive to maintain suitable environmental quality and must work toward continuous improvement of personnel operations and environmental control. In general, fewer personnel involved in aseptic processing and monitoring, along with reduction in interventions, reduces risk from microbial contamination.
APPENDIX
Airborne Particulate Count (also referred to as Total Particulate Count): The total number of particles of a given size per unit volume of air.
Airborne Viable Particulate Count (also referred to as Total Airborne Aerobic Microbial Count): The recovered number of colony-forming units (cfu) per unit volume of air.
Air Changes: The frequency per unit of time (minutes, hours, etc.) that the air within a controlled environment is replaced. The air can be recirculated partially or totally replaced.
Air Sampler: Devices or equipment used to sample a measured amount of air in a specified time to quantitate the particulate or microbiological status of air in the controlled environment.
Aseptic: Technically, the absence of microorganisms, but in aseptic processing this refers to methods and operations that minimize microbial contamination in environments where sterilized product and components are filled and/or assembled.
Aseptic Processing: An operation in which the product is assembled or filled into its primary package in an ISO 5 or better environment and under conditions that minimize the risk of microbial contamination. The ultimate goal is to produce products that are as free as possible of microbial contamination.
Barrier System: Physical barriers installed within an aseptic processing room to provide partial separation between aseptically gowned personnel and critical areas subject to considerable contamination risk. Personnel access to the critical zone is largely unrestricted. It is subject to a high level disinfection.
Bioburden: Total number and identity of the predominant microorganisms detected in or on an article.
Clean Room: A room in which the concentration of airborne particles is controlled to meet a specified airborne particulate cleanliness Class. In addition, the concentration of microorganisms in the environment is monitored; each cleanliness Class defined is also assigned a microbial level for air, surface, and personnel gear.
Commissioning of a Controlled Environment: Certification by engineering and quality control that the environment has been built according to the specifications of the desired cleanliness Class and that, under conditions likely to be encountered under normal operating conditions (or worst-case conditions), it is capable of delivering an aseptic process. Commissioning includes media-fill runs and results of the environmental monitoring program.
Contamination Recovery Rate: The contamination recovery rate is the rate at which environmental samples are found to contain any level of contamination. For example, an incident rate of 1% would mean that only 1% of the samples taken have any contamination regardless of colony number.
Controlled Environment: Any area in an aseptic process system for which airborne particulate and microorganism levels are controlled to specific levels, appropriate to the activities conducted within that environment.
Corrective Action: Actions to be performed that are according to standard operating procedures and that are triggered when certain conditions are exceeded.
Critical Zone: Typically the entire area where product and the containers and closures are exposed in aseptic processing.
Detection Frequency: The frequency with which contamination is observed in an environment. Typically expressed as a percentage of samples in which contamination is observed per unit of time.
Environmental Isolates: Microorganisms that have been isolated from the environmental monitoring program.
Environmental Monitoring Program: Documented program implemented via standard operating procedures that describes in detail the methods and acceptance criteria for monitoring particulates and microorganisms in controlled environments (air, surface, personnel gear). The program includes sampling sites, frequency of sampling, and investigative and corrective actions.
Equipment Layout: Graphical representation of an aseptic processing system that denotes the relationship between and among equipment and personnel. This layout is used in the Risk Assessment Analysis to determine sampling site and frequency of sampling based on potential for microbiological contamination of the product/container/closure system. Changes must be assessed by responsible managers, since unauthorized changes in the layout for equipment or personnel stations could result in increase in the potential for contamination of the product/container/closure system.
Isolator for Aseptic Processing: An aseptic isolator is an enclosure that is over-pressurized with HEPA filtered air and is decontaminated using an automated system. When operated as a closed system, it uses only decontaminated interfaces or rapid transfer ports (RTPs) for materials transfer. After decontamination they can be operated in an open manner with the ingress and/or egress of materials through defined openings that have been designed and validated to preclude the transfer of contamination. It can be used for aseptic processing activities or for asepsis and containment simultaneously.
Material Flow: The flow of material and personnel entering controlled environments should follow a specified and documented pathway that has been chosen to reduce or minimize the potential for microbial contamination of the product/closure/container systems. Deviation from the prescribed flow could result in increase in the potential for microbial contamination. Material/personnel flow can be changed, but the consequences of the changes from a microbiological point of view should be assessed by responsible managers and must be authorized and documented.
Media Fill: Microbiological simulation of an aseptic process by the use of growth media processed in a manner similar to the processing of the product and with the same container/closure system being used.
Media Growth Promotion: Procedure that references Growth Promotion Test of Aerobes, Anerobes, and Fungi in Sterility Tests
Product Contact Areas: Areas and surfaces in a controlled environment that are in direct contact with either products, containers, or closures and the microbiological status of which can result in potential microbial contamination of the product/container/closure system.
Restricted Access Barrier System (RABS): An enclosure that relies on HEPA filtered air over-spill to maintain separation between aseptically gowned personnel and the operating environment. It is subject to a high level of disinfection prior to use in aseptic process. It uses decontaminated (where necessary) interfaces or RTPs for materials transfer. It allows for the ingress and/or egress of materials through defined openings that have been designed and validated to preclude the transfer of contamination. If opened subsequent to decontamination, its performance capability is adversely impacted.
Risk Assessment Analysis: Analysis of the identification of contamination potentials in controlled environments that establish priorities in terms of severity and frequency and that will develop methods and procedures that will eliminate, reduce, minimize, or mitigate their potential for microbial contamination of the product/container/closure system.
Sampling Plan: A documented plan that describes the procedures and methods for sampling a controlled environment; identifies the sampling sites, the sampling frequency, and number of samples; and describes the method of analysis and how to interpret the results.
Sampling Sites: Documented geographical location, within a controlled environment, where sampling for microbiological evaluation is taken. In general, sampling sites are selected because of their potential for product/container–closure contacts.
Standard Operating Procedures: Written procedures describing operations, testing, sampling, interpretation of results, and corrective actions that relate to the operations that are taking place in a controlled environment and auxiliary environments. Deviations from standard operating procedures should be noted and approved by responsible managers.
Sterile or Aseptic Field: In aseptic processing or in other controlled environments, it is the space at the level of or above open product containers, closures, or product itself, where the potential for microbial contamination is highest.
Sterility: Within the strictest definition of sterility, an article is deemed sterile when there is complete absence of viable microorganisms. Viable, for organisms, is defined as having the capacity to reproduce. Absolute sterility cannot be practically demonstrated because it is technically unfeasible to prove a negative absolute. Also, absolute sterility cannot be practically demonstrated without testing every article in a batch. Sterility is defined in probabilistic terms, where the likelihood of a contaminated article is acceptably remote.
Swabs for Microbiological Sampling: Devices used to remove microorganisms from irregular or regular surfaces for cultivation to identify the microbial population of the surface. A swab is generally composed of a stick with an absorbent tip that is moistened before sampling and is rubbed across a specified area of the sample surface. The swab is then rinsed in a sterile solution to suspend the microorganisms, and the solution is transferred to growth medium for cultivation of the microbial population.
Trend Analysis: Data from a routine microbial environmental monitoring program that can be related to time, shift, facility, etc. This information is periodically evaluated to establish the status or pattern of that program to ascertain whether it is under adequate control. A trend analysis is used to facilitate decision-making for requalification of a controlled environment or for maintenance and sanitization schedules.
REFERENCES
Agalloco J, Akers J. Risk analysis for aseptic processing: The Akers-Agalloco method. Pharm Technol. 2005; 29(11):74–88.
Agalloco J, Akers J. The simplified Akers-Agalloco method for aseptic processing risk analysis. Pharm Technol. 2006; 31(7)60–72.
Akers, J. The proper role of environmental monitoring in aseptic processing. Am Pharm Rev. 2006; 9(4):24–28.
CDC, Healthcare Infection Control Advisory Committee. Guidelines for environmental control in healthcare facilities. MMWR 2003; 52(No. RR-10):1–42.
Favero MS, Puleo JR, Marshall JH, Oxborrow GS. Microbiological sampling of surfaces. J Appl Bacteriol. 1968; 31:336–346.
Hussong D, Madsen R. Analysis of environmental microbiology data from clean room samples. Pharm Technol. 2004; Aseptic Processing Suppl:10–15.
International Organization for Standardization (ISO). 14644-1, Clean rooms and associated environments, part 1: classification of air cleanliness. Geneva: ISO; 1999.
International Organization for Standardization (ISO). 14644-2, Clean rooms and associated environments, part 2: specifications for testing and monitoring to prove continued compliance with 14644 part 1. Geneva: ISO; 2000.
Jensen PA, Todd WF, Davis GN, Scarpino PV. Evaluation of eight bioaerosol samplers challenged with aerosol of free bacteria. Am Ind Hyg Assoc J. 1992; 53:660–667.
Ljungqvist B. Active sampling of airborne viable particulate in controlled environments: a comparative study of common instruments. Eur J Parenter Sci. 1998; 3:59–62.
Ljungqvist B, Reinmüller B. Interaction between air movements and the dispersion of contaminants: clean zones with unidirectional air flow. J Parenter Sci Technol. 1993; 47(2):60–69.
Ljungqvist B, Reinmüller B. Airborne viable particles and total number of airborne particles: comparative studies of active air sampling. PDA J Sci Technol. 2000; 54:112–116.
Maruyama M, Matsuoka T, Deguchi M, Akers J. The application of robotics to aseptic surface monitoring. Pharm Technol. 2007; 32(7):40–44.
Process simulation testing for sterile bulk pharmaceutical chemicals. PDA Technical Report No. 28. J Parenter Sci Technol. 1998; 52 S3.
Reinmüller B. Dispersion and risk assessment of airborne contaminants in pharmaceutical cleanrooms. Building Serv Eng Bull (Sweden). 2001; Bulletin No. 56.
Stewart SL, Grinshpun SA, Willeke K, Terzieva S, Ulevicius V, Donnelly J. Effect of impact stress on microbial recovery on an agar surface. Appl Environ Micro. 1995; 61:1232–1239.
Whyte W. Reduction of microbial dispersion by clothing. J Parenter Sci Technol. 1985; 39(1):51–60. USP35
USP35
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
|---|---|---|
| General Chapter | Radhakrishna S Tirumalai, Ph.D.
Principal Scientific Liaison 1-301-816-8339 |
(GCM2010) General Chapters - Microbiology |
USP35–NF30 Page 697
Pharmacopeial Forum: Volume No. 36(6) Page 1688