Ramipril
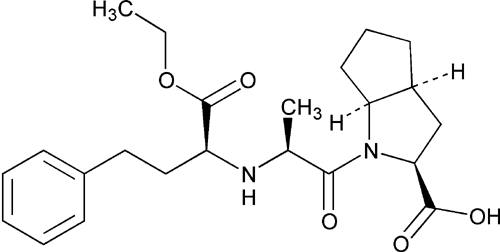
Cyclopenta[b]pyrrole-2-carboxylic acid, 1-[2-[[1-(ethoxycarbonyl)-3-phenylpropyl]amino]-1-oxopropyl]octahydro-, [2S-[1[R*(R*)],2
(2S,3aS,6aS)-1-[(S)-N-[(S)-1-Carboxy-3-phenylpropyl]alanyl]octahydrocyclopenta[b]pyrrole-2-carboxylic acid, 1-ethyl ester
» Ramipril contains not less than 98.0 percent and not more than 102.0 percent of C23H32N2O5, calculated on the dried basis.
Packaging and storage—
Preserve in tight containers.
USP Reference standards  11
11 —
—
USP Ramipril RS.
USP Ramipril Related Compound A RS.
USP Ramipril Related Compound B RS.
USP Ramipril Related Compound C RS.
USP Ramipril Related Compound D RS.
USP Ramipril RS.
USP Ramipril Related Compound A RS.
USP Ramipril Related Compound B RS.
USP Ramipril Related Compound C RS.
USP Ramipril Related Compound D RS.
Specific rotation 781S:
between +32.0 and +38.0, determined at 20.
and +38.0, determined at 20.
Test solution:
10 mg per mL, in 0.1 M methanolic hydrochloric acid.
Loss on drying 731—
Dry it in vacuum at a pressure not exceeding 5 mm of mercury at 60 for 6 hours: it loses not more than 0.2% of its weight.
Residue on ignition 281:
not more than 0.1%.
Limit of palladium—
Diluent—
Prepare a mixture of water and nitric acid (997:3).
Standard stock solution—
Transfer about 50 mg of palladium metal, accurately weighed, to a 100-mL volumetric flask, dissolve in 9 mL of hydrochloric acid, and dilute with water to volume.
Standard solutions—
Dilute the Standard stock solution quantitatively, and stepwise if necessary, with Diluent to obtain solutions containing 0.02, 0.03, and 0.05 µg of palladium per mL.
Test solution—
Transfer about 200 mg of Ramipril, accurately weighed, to a 100-mL volumetric flask, and dissolve in and dilute with Diluent to volume.
Blank solution—
Transfer about 150 mg of magnesium nitrate to a 100-mL volumetric flask, and dissolve in and dilute with Diluent to volume.
Procedure—
Concomitantly determine the absorbances of equal volumes of the Standard solutions and the Test solution (about 20 µL), at the palladium emission line at 247.6 nm, with a suitable atomic absorption spectrophotometer (see Spectrophotometry and Light-Scattering 851) equipped with a palladium hollow-cathode lamp, using a 10-µL injection of Blank solution as the blank. Plot the absorbances of the Standard solutions versus concentration, in µg per mL, of palladium, and draw the straight line best fitting the three plotted points. From the graph so obtained, determine the concentration, CP , in µg per mL, of palladium in the Test solution. Calculate the percentage of palladium in the portion of Ramipril taken by the formula:
0.1CP / CR,
in which CR is the concentration, in mg per mL, of Ramipril taken to prepare the Test solution. The limit is 0.002%.
Related compounds—
Solution A—
Dissolve 2.0 g of sodium perchlorate in a mixture of 800 mL of water and 0.5 mL of triethylamine, adjust with phosphoric acid to a pH of about 3.6 ± 0.1, add 200 mL of acetonitrile, and mix.
Solution B—
Dissolve 2.0 g of sodium perchlorate in a mixture of 300 mL of water and 0.5 mL of triethylamine, adjust with phosphoric acid to a pH of about 2.6 ± 0.1, add 700 mL of acetonitrile, and mix.
Mobile phase—
Use variable filtered and degassed mixtures of Solution A and Solution B as directed for Chromatographic system. Make adjustments if necessary (see System Suitability under Chromatography 621).
Test solution—
Transfer about 25 mg of Ramipril, accurately weighed, to a 25-mL volumetric flask, dissolve in and dilute with Solution A to volume, and mix. [note—Keep the Test solution cold until injected.]
Resolution solution—
Dissolve a quantity of USP Ramipril RS, USP Ramipril Related Compound A RS, USP Ramipril Related Compound B RS, USP Ramipril Related Compound C RS, and USP Ramipril Related Compound D RS in Solution B to obtain a solution with a concentration of about 0.5 mg of each per mL.
Standard solution—
Dissolve an accurately weighed quantity of USP Ramipril RS in Solution B, and dilute quantitatively, and stepwise if necessary, with Solution B to obtain a solution having a known concentration of about 0.005 mg per mL.
Chromatographic system (see Chromatography 621)—
The liquid chromatograph is equipped with a 210-nm detector and a 4.0-mm × 25-cm column that contains 3-µm packing L1, and is maintained at a temperature of 65. The flow rate is about 1 mL per minute. The chromatograph is programmed as follows.
| Time (minutes) |
Solution A
(%) |
Solution B
(%) |
Elution |
| 0–6 | 90 | 10 | isocratic |
| 6–7 | 90®75 | 10®25 | linear gradient |
| 7–20 | 75®65 | 25®35 | linear gradient |
| 20–30 | 65®25 | 35®75 | linear gradient |
| 30–40 | 25 | 75 | isocratic |
| 40–45 | 25®90 | 75®10 | linear gradient |
| 45–55 | 90 | 10 | isocratic |
note—Make adjustments at the 75:25 ratio stage, if necessary, to achieve elution of ramipril between 16 and 19 minutes after injection of the Standard solution.
Chromatograph the Resolution solution, and record the peak responses as directed for Procedure: the resolution, R, between ramipril related compound A and ramipril is not less than 3.0. Similarly chromatograph the Test solution, and record the peak responses as directed for Procedure: the retention time for ramipril is between 16 and 19 minutes; and the tailing factor for the ramipril peak is between 0.8 and 2.0. Chromatograph the Standard solution, and record the peak responses as directed for Procedure: the relative standard deviation for replicate injections is not more than 5.0%. [note—The relative retention times are about 0.8 for ramipril related compound A, 1.0 for ramipril, 1.3 for ramipril related compound B, 1.5 for ramipril related compound C, and 1.6 for ramipril related compound D.]
Procedure—
Separately inject equal volumes (about 10 µL) of the Test solution and the Standard solution into the chromatograph, record the chromatograms, and measure the peak response for ramipril obtained from the Standard solution and the responses of all the peaks, other than the ramipril peak, obtained from the Test solution. Calculate the percentage of each related compound and unknown impurity in the portion of Ramipril taken by the formula:
100F(CS / CT)(ri / rS)
in which F is the relative response factor for the related compound, which is 2.4 for ramipril related compound C, and 1.0 for all other individual impurities; CS is the concentration, in mg per mL, of USP Ramipril RS in the Standard solution; CT is the concentration, in mg per mL, of ramipril in the Test solution; ri is the peak response for each individual peak obtained from the Test solution; and rS is the ramipril peak response obtained from the Standard solution: not more than 0.5% of ramipril related compound A, ramipril related compound B, ramipril related compound C, or ramipril related compound D is found; not more than 0.1% of any other individual impurity is found; and not more than 1.0% of total impurities is found.
Assay—
Sodium dodecyl sulfate solution—
Prepare a 0.1% solution of sodium dodecyl sulfate. Adjust with phosphoric acid to a pH of 2.4 ± 0.1, filter, and degas.
Mobile phase—
Prepare a mixture of Sodium dodecyl sulfate solution and acetonitrile (55:45). Adjust with phosphoric acid to a pH of 2.75 ± 0.1, filter, and degas. Make adjustments if necessary (see System Suitability under Chromatography 621).
System suitability preparation—
Dissolve accurately weighed quantities of USP Ramipril RS and USP Ramipril Related Compound A RS in Mobile phase to obtain a solution having known concentrations of about 0.2 mg per mL and 0.01 mg per mL, respectively.
Standard preparation—
Dissolve an accurately weighed quantity of USP Ramipril RS in Mobile phase to obtain a solution having a known concentration of about 0.2 mg per mL.
Assay preparation—
Transfer about 100 mg of Ramipril, accurately weighed, to a 100-mL volumetric flask, dissolve in about 10 mL of acetonitrile, dilute with Mobile phase to volume, and mix. Pipet about 10 mL of this stock solution to a 50-mL volumetric flask, dilute with Mobile phase to volume, and mix.
Chromatographic system (see Chromatography 621)—
The liquid chromatograph is equipped with a 210-nm detector and a 4.6-mm × 15-cm column that contains packing L1. The flow rate is about 1.8 mL per minute. Chromatograph the System suitability preparation, and record the peak responses as directed for Procedure: the resolution, R, between ramipril and ramipril related compound A is not less than 2.0; the column efficiency determined from the ramipril peak is not less than 4000 theoretical plates; and the relative standard deviation for replicate injections determined from the ramipril peak is not more than 1.0%.
Procedure—
Separately inject equal volumes (about 20 µL) of the Standard preparation and the Assay preparation into the chromatograph, record the chromatograms, and measure the responses for all of the peaks. Calculate the quantity, in mg, of C23H32N2O5 in the portion of Ramipril taken by the formula:
500C(rU / rS)
in which C is the concentration, in mg per mL, of USP Ramipril RS in the Standard preparation; and rU and rS are the peak responses obtained from the Assay preparation and the Standard preparation, respectively.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
Chromatographic Column—
| Topic/Question | Contact | Expert Committee |
| Monograph | Sujatha Ramakrishna, Ph.D.
Scientist 1-301-816-8349 |
(MDCV05) Monograph Development-Cardiovascular |
| Reference Standards | Lili Wang, Technical Services Scientist 1-301-816-8129 RSTech@usp.org |
USP32–NF27 Page 3474
Pharmacopeial Forum: Volume No. 31(3) Page 787
Chromatographic columns text is not derived from, and not part of, USP 32 or NF 27.