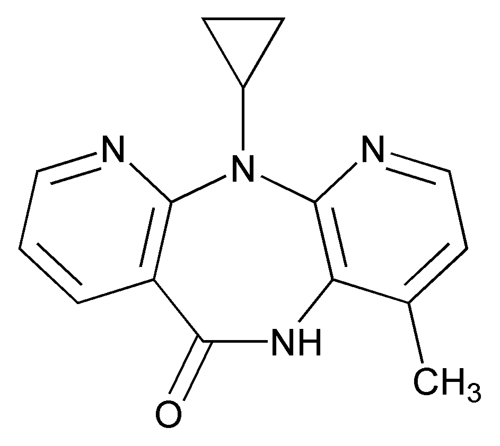
Nevirapine Oral Suspension
» Nevirapine Oral Suspension contains not less than 90.0 percent and not more than 110.0 percent of the labeled amount of nevirapine (C15H14N4O).
Packaging and storage—
Preserve in well-closed containers. Store at 25 , excursions permitted between 15 and 30.
, excursions permitted between 15 and 30.
USP Reference standards  11
11 —
—
USP Nevirapine Anhydrous RS .
.
USP Nevirapine Related Compund A RS. USP Nevirapine Related Compound B.
USP Nevirapine Anhydrous RS
 .
.
USP Nevirapine Related Compund A RS. USP Nevirapine Related Compound B.
Identification—
A:
Thin-Layer Chromatographic Identification Test 201—
Ferric chloride–potassium ferricyanide reagent—
Dissolve 1.35 g of ferric chloride in 25 mL of water. Dissolve 1.64 g of potassium ferricyanide in 25 mL of water. Mix the two solutions immediately before use.
Test solution—
Transfer a volume of Oral Suspension, equivalent to about 10 mg of nevirapine, to an 8-mL glass stoppered tube. Pipet 2.0 mL of chloroform into the tube, and shake. Allow the two phases to separate, and then using a disposable glass Pasteur pipet, remove some of the organic layer from the bottom, and transfer to another container.
Standard solution—
Dissolve a suitable quantity of USP Nevirapine Anhydrous RS in chloroform to obtain a solution having a known concentration of about 5 mg per mL.
Procedure—
Separately apply 5-µL portions of the Test solution and the Standard solution to a thin-layer chromatographic plate (see Chromatography 621) coated with a 0.25-mm layer of chromatographic silica gel 60 F254. Allow the spots to dry, and develop the chromatogram in a chamber saturated with a solvent system consisting of a mixture of ethyl acetate, isopropanol, and concentrated ammonium hydroxide (18:2:0.1) until the solvent front has moved about 6 to 7 cm from the point of application. Remove the plate from the chamber, mark the solvent front, and dry. Examine the chromatograms under UV light at 254 nm, and outline the spots with a soft pencil. Spray the plate with Ferric chloride–potassium ferricyanide reagent: the RF value (approximately 0.4 to 0.5) of the principal blue spot under UV and after spraying, obtained from the Test solution, corresponds to that obtained from the Standard solution.
B:
The retention time of the major peak in the chromatogram of the Assay preparation corresponds to that in the chromatogram of the Standard preparation, as obtained in the Assay.
Microbial enumeration tests 61 and Tests for specified microorganisms 62—
It meets the requirements of the tests for absence of Escherichia coli. The total aerobic microbial count does not exceed 100 cfu per mL, and the total combined molds and yeasts count does not exceed 50 cfu per mL.
Dissolution 711—
Medium:
0.1 N hydrochloric acid; 900 mL.
Apparatus 2:
25 rpm.
Time:
45 minutes.
Determine the amount of C15H14N4O dissolved by employing the following method.
Diluent:
a mixture of dehydrated alcohol and water (1:1).
Mobile phase—
Prepare a filtered and degassed mixture of water and acetonitrile (77:23). Make adjustments if necessary (see System Suitability under Chromatography 621).
System suitability solution—
Transfer about 10 mg of USP Nevirapine Anhydrous RS and 15 mg of methylparaben into a 250-mL volumetric flask. Dissolve in approximately 2 mL of Diluent, and dilute with Medium to volume.
Standard solution—
Transfer about 28 mg of USP Nevirapine Anhydrous RS, accurately weighed, into a 500-mL volumetric flask. Add 2 mL of Diluent, and sonicate for about 1 minute. Note that the Standard will not be completely dissolved at this point. Dilute with Medium to volume, and visually examine the solution to ensure that the Standard is completely dissolved. The final concentration is about 0.056 mg of nevirapine per mL.
Test solution—
For sample mixing, gently shake the bottle for approximately 10 seconds by inverting it slowly and rotating it from side to side. The sample should be free of air bubbles. Do not sonicate the sample. Using a 1- to 10-mL suitable positive displacement pipet set at 5 mL, withdraw the equivalent of 50 mg of nevirapine. Remove excess Oral Suspension by wiping the outside of the tip carefully so as not to touch the opening of the tip. Introduce the sample into the dissolution vessel over a 1- to 2-second time period by immersing the tip of the pipet midway between the paddle and the side of the vessel, approximately 1 cm below the meniscus. Similarly dispense the Oral Suspension into the other vessels. At 45 minutes, withdraw 5 mL of the solution under test, and pass through a 0.45-µm nylon filter, discarding the first 2 mL.
Chromatographic system (see Chromatography 621)—
The chromatograph is equipped with a 214-nm detector, a 3.9-mm × 20-mm guard column that contains packing L1, and a 3.9-mm × 15-cm analytical column that contains 5-µm packing L1. The flow rate is about 1.0 mL per minute. Chromatograph the System suitability solution, and record the peak responses as directed for Procedure: the resolution, R, between nevirapine and methylparaben is not less than 5.0; and the tailing factor for the nevirapine peak is not more than 1.8. Chromatograph the Standard solution, and record the peak responses as directed for Procedure: the relative standard deviation for replicate injections is not more than 2.0%.
Procedure—
Separately inject equal volumes (about 10 µL) of the Standard solution and the Test solution into the chromatograph, record the chromatograms for at least 14 minutes, and measure the responses for the nevirapine peaks. Calculate the percentage of C15H14N4O dissolved by the formula:
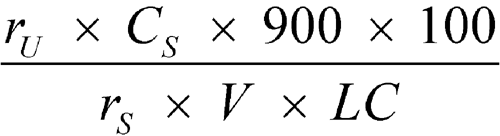 in which rU and rS are the peak responses obtained from the Test solution and the Standard solution, respectively; CS is the concentration, in mg per mL, of USP Nevirapine Anhydrous RS in the Standard solution; 900 is the volume, in mL, of the Medium; 100 is the conversion factor to percentage; V is the volume, in mL, of Oral Suspension taken; and LC is the Oral Suspension label claim, in mg per mL.
in which rU and rS are the peak responses obtained from the Test solution and the Standard solution, respectively; CS is the concentration, in mg per mL, of USP Nevirapine Anhydrous RS in the Standard solution; 900 is the volume, in mL, of the Medium; 100 is the conversion factor to percentage; V is the volume, in mL, of Oral Suspension taken; and LC is the Oral Suspension label claim, in mg per mL.
Tolerances—
Not less than 80% (Q) of the labeled amount of C15H14N4O is dissolved in 45 minutes.
Related compounds—
Potassium phosphate buffer, Solution A, Solution B, Mobile phase, and Diluent—
Proceed as directed in the Assay.
System suitability solution—
Proceed as directed in the Assay.
Standard stock solution—
Use the Standard stock preparation, prepared as directed in the Assay.
Standard solution—
Dilute the Standard stock solution quantitatively with Diluent to obtain a solution having a known concentration of about 0.3 µg of nevirapine per mL.
Weight determination—
Use the weight obtained as directed for Weight determination in the Assay.
Test solution—
Use the Assay preparation, prepared as directed in the Assay.
Chromatographic system (see Chromatography 621)—
Proceed as directed in the Assay: the relative standard deviation for replicate injections of the Standard solution is not more than 10.0%.
Procedure—
Separately inject equal volumes (about 20 µL) of the Standard solution and the Test solution into the chromatograph, record the chromatograms, and measure the peak responses. Calculate the percentage of each unknown impurity in the portion of Oral Suspension taken by the formula:
200(C/WU)(WA /5)(rU / rS)(100/L)
in which C is the concentration, in mg per mL, of USP Nevirapine Anhydrous RS in the Standard solution; WU is the sample weight, in g, taken to prepare the Test solution; WA is the weight, in g, of 5 mL of Oral Suspension, obtained as directed for Weight determination; L is the labeled amount, in mg per mL, of nevirapine in the Oral Suspension; rU is the peak response obtained for each impurity in the Test solution; and rS is the peak response for nevirapine in the Standard solution. Not more than 0.1% of any individual unknown impurity is found; and not more than 0.2% of total impurities is found. [note—The excipients and their degradation products should not be included in the determination of impurities.]
Assay—
Diluent—
Prepare a mixture of water and methanol (80:20).
Potassium phosphate buffer—
Dissolve 13.6 g of monobasic potassium phosphate in approximately 1900 mL of water, and adjust with phosphoric acid to a pH of 3.0. Transfer to a 2000-mL volumetric flask, and dilute with water to volume. Mix, filter, and degas.
Solution A:
a mixture of Potassium phosphate buffer and acetonitrile (97:3).
Solution B:
a mixture of Potassium phosphate buffer and acetonitrile (76:24).
Mobile phase—
Use variable mixtures of Solution A and Solution B as directed for Chromatographic system. Make adjustments if necessary (see System Suitability under Chromatography 621).
Standard stock preparation—
Transfer about 50 mg of USP Nevirapine Anhydrous RS, accurately weighed, into a 50-mL volumetric flask. Add 20 mL of methanol, and sonicate with intermittent swirling until the sample dissolves. Add water to about 1 cm below the meniscus, cool to room temperature, and dilute with water to volume. The concentration is about 1 mg of nevirapine per mL.
Standard preparation—
Dilute the Standard stock preparation quantitatively with Diluent to obtain a solution having a known concentration of about 0.3 mg of nevirapine per mL.
Stock impurity preparation—
Transfer about 3 mg of USP Nevirapine Related Compound A RS and 3 mg of USP Nevirapine Related Compound B RS, accurately weighed, into a 100-mL volumetric flask, add 20 mL of methanol, and sonicate to dissolve. Add water to about 1 cm below the meniscus, cool to room temperature, dilute with water to volume, and mix.
System suitability solution—
Transfer 15.0 mL of Standard stock preparation and 2.0 mL of Stock impurity preparation into a 50-mL volumetric flask, dilute with Diluent to volume, and mix.
Weight determination—
Using a 1- to 10-mL suitable pipet and a positive displacement tip, withdraw 5.0 mL of the Oral Suspension. The sample should be free of air bubbles. Dispense into a tared vial, and record the weight of the Oral Suspension to ±0.1 mg.
Assay preparation—
Using a 1- to 10-mL suitable pipet and a positive displacement tip, withdraw the equivalent of 60 mg of nevirapine. The sample should be free of air bubbles. Remove the excess Oral Suspension by wiping the outside of the tip carefully so as not to touch the opening of the tip, and deliver the sample into a 200-mL tared volumetric flask. Record the sample weight to the nearest ±0.1 mg. Add 40 mL of methanol, and sonicate for about 5 minutes with intermittent swirling. Add water to about 1 cm below the meniscus. Do not shake the flask. Allow the solution to attain room temperature, and dilute with water to volume. Shake the flask gently, and allow to stand for about 5 minutes.
Chromatographic system 621—
The liquid chromatograph is equipped with a 254-nm detector, a 4.6-mm × 12.5-mm guard column that contains 5-µm packing L10, and a 4.6-mm × 15-cm analytical column that contains 3.5-µm packing L10. The column temperature is maintained at 35. The flow rate is about 1.5 mL per minute. The chromatograph is programmed as follows.
Chromatograph the System suitability solution (about 20 µL), and record the peak responses as directed for Procedure: the resolution, R, between nevirapine and nevirapine related compound A is not less than 3.0, and the resolution, R, between nevirapine and nevirapine related compound B is not less than 1.7; and the tailing factor for the nevirapine peak is not more than 1.5. Chromatograph the Standard preparation, and record the peak responses as directed for Procedure: the relative standard deviation for replicate injections is not more than 2.0%.
| Time (minutes) |
Solution A
(%) |
Solution B
(%) |
Elution |
| 0–1 | 100 | 0 | isocratic |
| 1–31 | 100®0 | 0®100 | linear gradient |
| 31–32 | 0®100 | 100®0 | linear gradient |
| 32–42 | 100 | 0 | equilibration |
Procedure—
Separately inject equal volumes (about 20 µL) of the Standard preparation and the Assay preparation into the chromatograph, record the chromatograms, and measure the responses for the nevirapine peak. Calculate the quantity, in mg, of nevirapine (C15H14N4O) in each mL of the Oral Suspension taken by the formula:
200(C/WU)(WA /5)(rU / rS)
in which C is the concentration, in mg per mL, of USP Nevirapine Anhydrous RS in the Standard preparation; WU is the sample weight, in g, of Oral Suspension taken to prepare the Assay preparation; WA is the weight, in g, of 5 mL of Oral Suspension obtained as directed for Weight determination; and rU and rS are the peak responses obtained from the Assay preparation and the Standard preparation, respectively.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
Chromatographic Column—
| Topic/Question | Contact | Expert Committee |
| Monograph | Behnam Davani, Ph.D., M.B.A.
Senior Scientist 1-301-816-8394 |
(MDAA05) Monograph Development-Antivirals and Antimicrobials |
| Reference Standards | Lili Wang, Technical Services Scientist 1-301-816-8129 RSTech@usp.org |
|
| Radhakrishna S Tirumalai, Ph.D.
Senior Scientist 1-301-816-8339 |
(MSA05) Microbiology and Sterility Assurance | |
| Radhakrishna S Tirumalai, Ph.D.
Senior Scientist 1-301-816-8339 |
(MSA05) Microbiology and Sterility Assurance | |
| Margareth R.C. Marques, Ph.D.
Senior Scientist 1-301-816-8106 |
(BPC05) Biopharmaceutics05 |
USP32–NF27 Page 3073
Pharmacopeial Forum: Volume No. 32(4) Page 1090
Chromatographic columns text is not derived from, and not part of, USP 32 or NF 27.