Cefpiramide
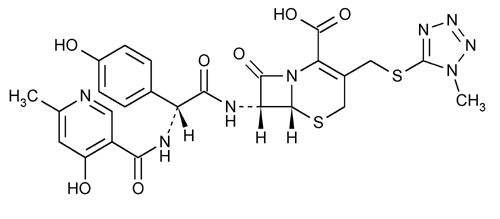
5-Thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid, 7-[[[[(4-hydroxy-6-methyl-3-pyridinyl)carbonyl]amino](4-hydroxyphenyl)acetyl]amino]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-8-oxo, [6R-[6
(6R,7R)-7-[(R)-2-(4-Hydroxy-6-methylnicotinamido)-2-(p-hydroxyphenyl)acetamido]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid
» Cefpiramide contains not less than 974 µg and not more than 1026 µg of C25H24N8O7S2 per mg, calculated on the anhydrous basis.
Packaging and storage—
Preserve in tight containers.
Labeling—
Where it is intended for use in preparing injectable dosage forms, the label states that it is sterile or must be subjected for further processing during the preparation of injectable dosage forms.
Identification—
B:
The retention time of the major peak in the chromatogram of the Assay preparation corresponds to that in the chromatogram of the Standard preparation, as obtained in the Assay.
Crystallinity  695
695 :
meets the requirements.
:
meets the requirements.
pH 791:
between 3.0 and 5.0, in a suspension (1 in 200).
Water, Method I 921:
not more than 9.0%.
Related compounds—
pH 7.5 Buffer—
Dissolve 4.08 g of monobasic potassium phosphate in 800 mL of water, adjust with 1 N sodium hydroxide to a pH of 7.5 ± 0.1, dilute with water to 1000 mL, and mix.
Mobile phase—
Prepare a suitable filtered and degassed mixture of pH 7.5 Buffer and methanol (750:250). Make adjustments if necessary (see System Suitability under Chromatography 621).
Resolution solution and Chromatographic system—
Proceed as directed in the Assay.
Standard solution—
Transfer about 15 mg of sodium 5-mercapto-1-methyltetrazole and 25 mg of USP Cefpiramide RS, both accurately weighed, to a 100-mL volumetric flask, dissolve in and dilute with pH 7.5 Buffer to volume, and mix. Transfer 2.0 mL of this solution to a second 100-mL volumetric flask, dilute with Mobile phase to volume, and mix. This solution contains about 3 µg of sodium 5-mercapto-1-methyltetrazole and about 5 µg of USP Cefpiramide RS per mL.
Test solution—
Transfer about 25 mg of Cefpiramide, accurately weighed, to a 50-mL volumetric flask, dissolve in and dilute with Mobile phase to volume, and mix.
Procedure—
Separately inject equal volumes (about 20 µL) of the Standard solution and the Test solution into the chromatograph, record the chromatograms, and measure the areas for all of the peaks. Calculate the percentage of 5-mercapto-1-methyltetrazole in the portion of Cefpiramide taken by the formula:
921, but prepare the Test Preparation as follows. Transfer about 100 mg of sodium 5-mercapto-1-methyltetrazole, accurately weighed, to a stoppered centrifuge tube. Add 10.0 mL of a solution of N-ethylmaleimide in methanol (4 in 100), and sonicate for 15 minutes. Titrate 5.0 mL of this Test Preparation.) in the sodium 5-mercapto-1-methyltetrazole taken to prepare the Standard solution; W is the weight, in mg, of Cefpiramide taken to prepare the Test solution; and rU and rS are the 5-mercapto-1-methyltetrazole peak area responses obtained from the Test solution and the Standard solution, respectively: not more than 0.7% of 5-mercapto-1-methyltetrazole is found. Calculate the percentage of each other impurity in the portion of Cefpiramide taken by the formula:
(115.14/138.13)[5Ct (100  w)/100W](rU / rS)
w)/100W](rU / rS)
in which 115.14 and 138.13 are the molecular weights of 5-mercapto-1-methyltetrazole and anhydrous sodium 5-mercapto-1-methyltetrazole, respectively; Ct is the concentration, in µg per mL, of sodium 5-mercapto-1-methyltetrazole in the Standard solution; w is the percentage of water, determined by the titrimetric method (See Water Determination 5(CE/1000W)(rU / rS)
in which C is the concentration, in µg per mL, of USP Cefpiramide RS in the Standard solution; E is the designated cefpiramide content, in µg per mg, of USP Cefpiramide RS; W is the weight, in mg, of Cefpiramide taken to prepare the Test solution; rU is the response of each other impurity obtained from the Test solution; and rS is the cefpiramide peak response obtained from the Standard solution: not more than 0.7% of any other individual impurity is found. The total of the percentages of 5-mercapto-1-methyltetrazole and all other impurities is not more than 2.0%.
Other requirements—
Where the label states that Cefpiramide is sterile, it meets the requirements for Sterility and Pyrogen under Cefpiramide for Injection. Where the label states that Cefpiramide must be subjected to further processing during the preparation of injectable dosage forms, it meets the requirements for Pyrogen under Cefpiramide for Injection.
Assay—
pH 6.8 Buffer—
Dissolve 1.36 g of monobasic potassium phosphate in 900 mL of water, adjust with 1 N sodium hydroxide to a pH of 6.8 ± 0.1, dilute with water to 1000 mL, and mix.
Mobile phase—
Prepare a suitable filtered and degassed mixture of pH 6.8 Buffer, acetonitrile, tetrahydrofuran, and methanol (900:20:60:20). Make adjustments if necessary (see System Suitability under Chromatography 621).
Resolution solution—
Prepare a solution of USP Cefpiramide RS in 0.01 N sodium hydroxide containing about 1 mg per mL. Heat this solution at 95 for 10 minutes. Mix 1 mL of this solution with 19 mL of Mobile phase. This solution contains a mixture of cefpiramide and cefpiramide lactone.
for 10 minutes. Mix 1 mL of this solution with 19 mL of Mobile phase. This solution contains a mixture of cefpiramide and cefpiramide lactone.
Standard preparation—
Dissolve an accurately weighed quantity of USP Cefpiramide RS in Mobile phase to obtain a solution having a known concentration of about 0.25 mg per mL.
Assay preparation—
Transfer about 50 mg of Cefpiramide, accurately weighed, to a 200-mL volumetric flask, dissolve in and dilute with Mobile phase to volume, and mix.
Chromatographic system (see Chromatography 621)—
The liquid chromatograph is equipped with a 254-nm detector and a 4.6-mm × 25-cm column that contains packing L1. The flow rate is about 1.5 mL per minute. Chromatograph the Resolution solution, and record the peak responses as directed for Procedure: the relative retention times are about 0.7 for cefpiramide and 1.0 for cefpiramide lactone; and the resolution between the cefpiramide lactone peak and the cefpiramide peak is not less than 5. Chromatograph the Standard preparation, and record the peak responses as directed for Procedure: the capacity factor, k¢, is between 2 and 5 and the column efficiency is not less than 1700 theoretical plates when calculated by the formula:
5.545(tr / Wh / 2)2
the tailing factor for the cefpiramide peak is not less than 0.95 and not more than 1.4; and the relative standard deviation for replicate injections is not more than 2.0%.
Procedure—
Separately inject equal volumes (about 20 µL) of the Standard preparation and the Assay preparation into the chromatograph, record the chromatograms, and measure the areas for the major peaks. Calculate the quantity, in µg, of C25H24N8O7S2 in each mg of the Cefpiramide taken by the formula:
200(CE/W)(rU / rS)
in which C is the concentration, in mg per mL, of USP Cefpiramide RS in the Standard preparation; E is the designated cefpiramide (C25H24N8O7S2) content, in µg per mg, of USP Cefpiramide RS; W is the weight, in mg, of the Cefpiramide taken to prepare the Assay preparation; and rU and rS are the cefpiramide peak responses obtained from the Assay preparation and the Standard preparation, respectively.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
Chromatographic Column—
| Topic/Question | Contact | Expert Committee |
| Monograph | Ahalya Wise, M.S.
Scientist 1-301-816-8161 |
(MDANT05) Monograph Development-Antibiotics |
| Reference Standards | Lili Wang, Technical Services Scientist 1-301-816-8129 RSTech@usp.org |
USP32–NF27 Page 1851
Pharmacopeial Forum: Volume No. 28(3) Page 747
Chromatographic columns text is not derived from, and not part of, USP 32 or NF 27.