This chapter provides guidelines for the validation of methods for the estimation of the number of viable microorganisms, for the detection of indicators or objectionable microorganisms, for the validation of microbiological methods used in antimicrobial effectiveness testing, and for the sterility testing of Pharmacopeial articles. It is generally understood that if a product possesses antimicrobial properties because of the presence of a specific preservative or because of its formulation, this antimicrobial property must be neutralized to recover viable microorganisms. This neutralization may be achieved by the use of a specific neutralizer, by dilution, by a combination of washing and dilution, or by any combination of these methods.
The tests under Antimicrobial Effectiveness Testing  51
51 , Sterility Tests 71, and Microbial Enumeration Tests 61 and Tests for Specified Microorganisms 62 require the validation of recovery methods. To ensure that the results of the tests are credible, neutralization of antimicrobial properties of the test solution is required before estimating the number of viable microorganisms.
, Sterility Tests 71, and Microbial Enumeration Tests 61 and Tests for Specified Microorganisms 62 require the validation of recovery methods. To ensure that the results of the tests are credible, neutralization of antimicrobial properties of the test solution is required before estimating the number of viable microorganisms.
INFLUENTIAL FACTORS
Several factors affect the measurement of a test solution's antimicrobial activity, and these must be considered in the validation design. They include the nature of the microorganisms used as challenge organisms, the preparation of the inoculum of challenge organisms, the specific conditions of the test, and the conditions of recovery. These factors also affect the validation of recovery methods for aqueous or nonaqueous products, irrespective of their antimicrobial properties; thus, all test methods should be validated with these factors in mind.
The nature of the challenge microorganism exerts a strong effect upon the response to the antimicrobial agent, and so upon the neutralization required for recovery. Represented among these organisms in compendial tests are Gram-positive bacteria, Gram-negative bacteria, yeasts, and molds. Each organism to be used in the test must be included in the validation.
The preparation of the inoculum of challenge microorganisms also affects the testing of products having antimicrobial properties. The growth and preparation of the challenge organism determines the physiological state of the cell. This state has a direct influence on the results of any test of antimicrobial efficacy. Microbial tests do not use individual cells; rather, populations of cells are harvested for study. The data generated from these studies are less variable if the cell populations are homogeneous. Liquid cultures or confluent growths on solid medium are best suited for reproducible culture preparation. The conditions of organism preparation and storage must be standardized for the neutralizer evaluation and should reflect the conditions of the antimicrobial assay.
The specific conditions of the test, including buffers used, water, light conditions, and temperature, must be reproduced in the validation study. All test conditions also should be standardized and performed in the validation study exactly as performed in the test.
The conditions of microbial recovery are among the most crucial in accurately estimating the number of microorganisms present in a test solution. The first consideration is the recovery medium used to support the growth of survivors. This concern is discussed in detail below. The second consideration is the incubation conditions. Optimal conditions for growth must be present to ensure complete growth and reproducible results.
METHODS OF NEUTRALIZING ANTIMICROBIAL PROPERTIES
Three common methods are used to neutralize antimicrobial properties of a product: (1) chemical inhibition, (2) dilution, and (3) filtration and washing.
Chemical Inhibition
Table 1 shows known neutralizers for a variety of chemical antimicrobial agents and the reported toxicity of some chemical neutralizers to specific microorganisms. However, despite potential toxicity, the convenience and quick action of chemical inhibitors encourage their use. Chemical inhibition of bactericides is the preferred method for the antimicrobial efficacy test. The potential of chemical inhibitors should be considered in the membrane filtration and the direct transfer sterility tests. Antibiotics may not be susceptible to neutralization by chemical means, but rather by enzymatic treatment (e.g., penicillinase). These enzymes may be used where required.
Table 1. Some Common Neutralizers for Chemical Biocides
| Neutralizer | Biocide Class | Potential Action of Biocides |
| Bisulfate | Glutaraldehyde, Mercurials | Non-Sporing Bacteria |
| Dilution | Phenolics, Alcohol, Aldehydes, Sorbate | — |
| Glycine | Aldehydes | Growing Cells |
| Lecithin | Quaternary Ammonium Compounds (QACs), Parabens, Bis-biguanides |
Bacteria |
| Mg+2 or Ca+2 ions | EDTA | — |
| Polysorbate | QACS, Iodine, Parabens | — |
| Thioglycollate | Mercurials | Staphylococci and Spores |
| Thiosulfate | Mercurials, Halogens, Aldehydes | Staphylococci |
Dilution
A second approach to neutralizing antimicrobial properties of a product is by dilution, because the concentration of a chemical bactericide exerts a large effect on its potency. The relationship between concentration and antimicrobial effect differs among bactericidal agents but is constant for a particular antimicrobial agent. This relationship is exponential in nature, with the general formula:
C 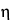 t = k
t = k
in which C is the concentration; t is the time required to kill a standard inoculum; k is a constant; and the concentration exponent, , is the slope of the plot of log t versus log C. Antimicrobial agents with high values are rapidly neutralized by dilution, whereas those with low values are not good candidates for neutralization by dilution.
Membrane Filtration
An approach that is often used, especially in sterility testing, is neutralization by membrane filtration. This approach relies upon the physical retention of the microorganism on the membrane filter, with the antimicrobial agent passing through the filter into the filtrate. The filter is then incubated for recovery of viable microorganisms. However, filtration alone may not remove sufficient quantities of the bactericidal agent to allow growth of surviving microorganisms. Adherence of residual antimicrobial agents to the filter membrane may cause growth inhibition. Filtration through a low-binding filter material, such as polyvinylidene difluoride, helps to minimize this growth inhibition. Additionally, the preservative may be diluted or flushed from the filter by rinsing with a benign fluid, such as diluting Fluid A (see Diluting and Rinsing Fluids for Membrane Filtration under Sterility Tests 71 for diluting fluid compositions). Chemical neutralizers in the rinsing fluid can ensure that any antimicrobial residue on the membrane does not interfere with the recovery of viable microorganisms.
VALIDATION OF NEUTRALIZATION METHODS—RECOVERY COMPARISONS
A validated method for neutralizing the antimicrobial properties of a product must meet two criteria: neutralizer efficacy and neutralizer toxicity. The validation study documents that the neutralization method employed is effective in inhibiting the antimicrobial properties of the product (neutralizer efficacy) without impairing the recovery of viable microorganisms (neutralizer toxicity). Validation protocols may meet these two criteria by comparing recovery results for treatment groups.
The first is the test group, in which the product is subjected to the neutralization method, then a low level of challenge microorganism [less than 100 colony-forming units (cfu)] is inoculated for recovery. The second is the peptone control group, in which the neutralization method is used with peptone, or diluting Fluid A (see Sterility Tests 71), as the test solution. The third is the viability group, in which the actual inoculum is used without exposure to the neutralization scheme. Similar recovery between the test group and the peptone group demonstrates adequate neutralizer efficacy; similar recovery between the peptone group and the viability group demonstrates adequate neutralizer toxicity.
In principle, the protocol must show that recovery of a low inoculum (less than 100 cfu) is not inhibited by the test sample and the neutralization method. Validation protocols may meet these two criteria by comparing recovery among three distinct test groups: (1) neutralized product with inoculum, (2) challenge inoculum control in buffered solution, and (3) inoculum in the absence of product or neutralizer. This can be established by directly comparing the result in the treated solution (1) to the inoculum (3) above. If the growth on the treated solution is not comparable to the growth on the inoculum group, it should be determined whether the neutralization method itself is toxic to the microorganisms.
Recovery on Agar Medium
In the tests under Antimicrobial Effectiveness Testing 51 and Microbial Enumeration Tests 61 and Tests for Specified Microorganisms 62, the number of viable challenge microorganisms in the product is estimated at various time intervals by calculating the concentration of cfu per mL by the plate count method. A design for validating neutralization would incorporate the treatment groups as described under Validation of Neutralization Methods—Recovery Comparisons. At least three independent replicates of the experiment should be performed, and each should demonstrate that the average number of cfu recovered from the challenge product is not less than 70% of that recovered from the inoculum control.
If a greater number of replicates is required in the validation study, the comparisons may be evaluated by transforming the numbers of cfu to their logarithmic values and analyzing the data statistically by the Student t test (pairwise comparisons) or by analysis of variance (ANOVA) (for comparing all groups). If ANOVA is used, and significant differences among the populations are determined, a test such as Dunnett's test may be used, with the peptone group used as the control group.
Recovery by Membrane Filtration
This validation follows the procedure described for Validation Test under Sterility Tests 71, with the exception of plating on solid medium to quantitate recovery. Three 100-mL rinses are assumed, but the volume and number of rinses are subject to validation. Each validation run should be performed independently at least three times.
In the test solution group, the product is filtered through the membrane filter, followed by two 100-mL portions of diluting-neutralizing fluid. After the second rinse has been filtered, a final 100-mL portion containing less than 100 cfu of the specific challenge microorganism is passed through the filter. This filter is then placed on the appropriate agar recovery medium and incubated for recovery.
The inoculum is directly plated onto the solid medium. It is possible that filtration will lead to reduced recovery of the challenge microorganism, either through inherent toxicity of the membrane or by adherence of the microorganism to the filtration vessel walls. A control group can be used to evaluate this component of membrane filtration validation. Diluting Fluid A is used as the dilution medium without exposing the filter to the product. After addition of the low-level inoculum to the final rinse, the filter is plated as above. Technique-specific loss of microorganisms can be estimated by comparing the recovery in the diluting Fluid A group to the inoculum count.
It is assumed in this discussion that the test sample can be filtered. If it is necessary to solubilize the test sample, the effects of the solubilization method on viable microorganisms must be determined. This situation can occur when testing ointments, suspensions, or other articles.
The method can be considered validated if the recovery rate in the three independent replicates is similar for the test solution and the diluting Fluid A control.
Recovery in Liquid Medium
It is assumed in Direct Inoculation of the Culture Medium in the section Test for Sterility of the Product to be Examined under Sterility Tests 71 that the recovery medium will allow for growth of all surviving microorganisms. The broth in that test must serve both to neutralize any antimicrobial properties of the test solution and to support the growth of the microorganisms. The treatment groups described under Validation of Neutralization Methods—Recovery Comparisons above can be used for validation of the recovery method, with the proportions of product and recovery medium varied to achieve adequate neutralization. The method can be considered validated if all groups show copious growth within 7 days for all microorganisms.
RECOVERY OF INJURED MICROORGANISMS
The validation studies described above use challenge microorganisms that have never been exposed to antimicrobial agents, and thus are not identical to organisms seen in antimicrobial effectiveness testing or when a sterility test is performed on a preserved product. If the use of alternative media is desired, the recovery of injured microorganisms should be addressed in the validation study. This may be done by directly comparing the recovery of each challenge microorganism on the preferred medium and on the alternative medium, after exposure to the product. This exposure should include at least two time periods showing survival of less than 100 cfu per mL, unless the rate of kill of the antimicrobial agent is such that no recovery is possible even if the microorganism is plated within minutes of exposure. This comparison should be performed at least three times. The alternative medium is validated if the recovery seen on that medium is no less than that seen on the preferred medium, within an error of 0.5 log units.
ESTIMATING THE NUMBER OF COLONY-FORMING UNITS
The accuracy of any estimate of viable cfu is affected by the number plated. As the number of viable cells plated increases, crowding effects decrease the accuracy of the count, reducing the estimate. As the number decreases, random error plays an increasing role in the estimate.
The accepted range for countable colonies on a standard agar plate is between 25 and 250 for most bacteria and Candida albicans. This range was established in the food industry for counting coliform bacteria in milk. This range is acceptable for compendial organisms, except for fungi. It is not optimal for counting all environmental isolates. The recommended counting range for Aspergillus niger is between 8 and 80 cfu per plate. The use of membrane filtration to recover challenge microorganisms, or the use of environmental isolates as challenge microorganisms in antimicrobial effectiveness testing, requires validation of the countable range. This validation may be performed by statistical comparison of estimated cfu from successive pairs in a dilution series. Prepare a suspension so that plating will provide approximately 1000 cfu per plate, and then dilute twofold to a theoretical concentration of approximately 1 cfu per plate. Plate all dilutions in the series in duplicate, and incubate for recovery under the conditions of the Antimicrobial Effectiveness Testing 51. Compare the estimates of cfu per mL from paired tubes in the dilution series by the formula:
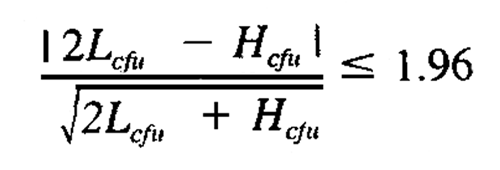
in which Lcfu is the number of colonies on the plate with the lower count (greater dilution), and Hcfu is the number of colonies on the plate with the higher count (lesser dilution). The estimates of the cfu per mL provided by Lcfu and Hcfu should agree within the limits of the formula with a critical value of 1.96. The upper limit of plate counts is then defined as the number (Hcfu) that reproducibly passes this test. This study should be independently repeated a sufficient number of times to establish an upper limit of cfu for the particular plating conditions.
There is a lower limit at which the ability of the antimicrobial effectiveness test to recover microorganisms becomes untenable. If the first plating is performed with 1 mL of a 10–1 dilution, cfu in the range of 1 to 10 per mL would not be seen. On this dilution plating, only the lower number of cfu may be reduced to 3, allowing as few survivors as 30 cfu per mL to be reported.
Lower counting thresholds for the greatest dilution plating in series must be justified. Numbers of colonies on a plate follow the Poisson distribution, so the variance of the mean value equals the mean value of counts. Therefore, as the mean number of cfu per plate becomes lower, the percentage error of the estimate increases (see Table 2). Three cfu per plate at the 10–1 dilution provide an estimate of 30 cfu per mL, with an error of 58% of the estimate.
Table 2. Error as a Percentage of Mean for Plate Counts
| cfu per
Plate |
Standard Error | Error as % of Mean |
| 30 | 5.48 | 18.3 |
| 29 | 5.39 | 18.6 |
| 28 | 5.29 | 18.9 |
| 27 | 5.20 | 19.2 |
| 26 | 5.10 | 19.6 |
| 25 | 5.00 | 20.0 |
| 24 | 4.90 | 20.4 |
| 23 | 4.80 | 20.9 |
| 22 | 4.69 | 21.3 |
| 21 | 4.58 | 21.8 |
| 20 | 4.47 | 22.4 |
| 19 | 4.36 | 22.9 |
| 18 | 4.24 | 23.6 |
| 17 | 4.12 | 24.3 |
| 16 | 4.00 | 25.0 |
| 15 | 3.87 | 25.8 |
| 14 | 3.74 | 26.7 |
| 13 | 3.61 | 27.7 |
| 12 | 3.46 | 28.9 |
| 11 | 3.32 | 30.2 |
| 10 | 3.16 | 31.6 |
| 9 | 3.00 | 33.3 |
| 8 | 2.83 | 35.4 |
| 7 | 2.65 | 37.8 |
| 6 | 2.45 | 40.8 |
| 5 | 2.24 | 44.7 |
| 4 | 2.00 | 50.0 |
| 3 | 1.73 | 57.7 |
| 2 | 1.41 | 70.7 |
| 1 | 1.00 | 100.0 |
Auxiliary Information— Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
| General Chapter | Radhakrishna S Tirumalai, Ph.D.
Senior Scientist 1-301-816-8339 |
(MSA05) Microbiology and Sterility Assurance |
USP32–NF27 Page 737