ANALYTICAL METHODOLOGIES
General Considerations
The complexity and scope of cell and gene therapy products is reflected in the wide range of analytical methods that are used to assess product quality. Approved cell and gene therapy products must comply with applicable sections of 21 CFR 211 and 21 CFR 610 to ensure their identity, dose, potency, purity, and safety. Specific guidance for the identification, development, and validation of analytical methodologies to support cell and virus bank characterization, final-product release, and stability studies is currently provided in the Center for Biologics Evaluation and Research (CBER) Points to Consider for Human and Somatic Cell and Gene Therapy (April 1998); under Validation of Compendial Procedures  1225
1225 ; and in the ICH guidelines entitled “Q2A Validation of Analytical Procedures”, “Q2B Validation of Analytical Procedures: Methodology”; and “Q6B Specification, Tests and Procedures for Biotechnological/Biological Products”. Most product-specific analytical methods for cell and gene therapy products have not been standardized. Even well-defined tests such as those described under Sterility Tests 71 may not be directly applicable to certain cell and gene therapy products. For some cell and gene therapy products, large quantities of clinical material may not be available during early clinical development. Some required tests (e.g., sterility) may have to be modified. Consultation with the regulatory authorities is advised.
; and in the ICH guidelines entitled “Q2A Validation of Analytical Procedures”, “Q2B Validation of Analytical Procedures: Methodology”; and “Q6B Specification, Tests and Procedures for Biotechnological/Biological Products”. Most product-specific analytical methods for cell and gene therapy products have not been standardized. Even well-defined tests such as those described under Sterility Tests 71 may not be directly applicable to certain cell and gene therapy products. For some cell and gene therapy products, large quantities of clinical material may not be available during early clinical development. Some required tests (e.g., sterility) may have to be modified. Consultation with the regulatory authorities is advised.
Table 6 provides an overview of the product-specific testing parameters for the biological component and general methods or approaches being used to satisfy the testing requirements for cell therapy products and for nonviral, viral, and antisense-oligonucleotide gene therapy products. The analysis of cell and gene therapy products relies heavily on biological assays but it also utilizes methodologies developed for biotechnology-derived products (see Biotechnology-Derived Articles 1045). Antisense-oligonucleotide products are regulated by the FDA Center for Drug Evaluation and Research (CDER) and rely heavily on physicochemical methods. The intent of this section is to outline the types of methods and their specific applications with regard to product characterization, stability, and release testing. Process validation may alleviate the need for certain specific lot-release tests. Development of appropriate reference materials and standards for cell therapy and viral and nonviral gene therapy products should be a part of product development. Reference materials should be fully characterized in order to provide continuity between standards over time. In the case of cellular products the reference material may be a surrogate tissue or simulated product.
Table 6. Analytical Tests for Cell and Gene Therapy Biological Products
Analytical Tests for Cell and Gene Therapy Biological Products
| Gene Therapy Products | |||
| Test | Cell Therapy Products | Viral | Nonviral and Antisense-Oligonucleotide |
| Identity of biological substance | Surface marker determination | Restriction enzyme map | Restriction enzyme map |
| Species | PCR | PCR | |
| Morphology | Immunoassay for expressed gene | Immunoassay for expressed gene | |
| Bioassay | Sequencing | Sequencing | |
| Biochemical marker | |||
| Dose | Viable cell number | Particle number | Plasmid-DNA weight |
| Enumeration of specific cell population | Transducing units (DNA hybridization assay) | Formulated-complex weight HPLC or capillary electrophores is assay using authenticated reference standard | |
| Total DNA | Total protein | ||
| Total protein | HPLC assay using authenticated reference standard | ||
| Potency | Viable cell number (cells intended for structural repair) | Function of expressed gene (induction of secondary effect and other bioassays) | Function of expressed gene (induction of secondary effect and other bioassays) |
| Bioassays: Colony-formation assay Function of expressed gene Induction of secondary effect (e.g., human leukocyte antigen (HLA) induction, secretion of cytokines, and up-regulation of surface marker) |
|||
| Purity | Percentage of viable cells | Residual host-cell DNA | Percentage of specific physical form (e.g., percentage supercoiled) |
| Percentage of transduced cells | Process contaminants (e.g., serum and cesium chloride) | Residual host-cell DNA | |
| Percentage of cells with specific surface marker | Residual helper virus | Residual RNA | |
| Process contaminants (e.g., serum) | Optical density ratio | Residual host-cell proteins | |
| Residual host-cell proteins | Residual solvents | ||
| Viral protein profile (HPLC assay for defective or immature particles) | Optical density ratio | ||
| Residual RNA | Process contaminants (e.g., cesium chloride and synthetic oligonucleotide by-products) | ||
| Safety | Mycoplasma | General safety | |
| Sterility | Mycoplasma | Mycoplasma | |
| Pyrogen and endotoxins | Sterility | Sterility | |
| Adventitious viruses | Pyrogen and endotoxins | Pyrogen and endotoxins | |
| Residual virus (for transfected cells) | Adventitious viruses | ||
| Replication-competent vector virus (transfected cells) | RCV | ||
Cell therapy products may require a rapid-release approach if they have a limited shelf life. The rapid-release approach is not usually applied to viral and nonviral gene therapy products because these products are sufficiently stable for completion of testing prior to release. Some formulated nonviral gene therapy products also have limited shelf lives. In such cases, the individual components are tested prior to release and the formulated complex is not tested. The formation and stability of the formulated nonviral gene therapy complex is established through validation studies during product development.
As specified in the CFR, product samples must be retained after product-release testing is completed. Additional samples may need to be retained if rapid-release strategies are employed, so that the product quality can be reassessed by alternative or traditional test methodologies if necessary.
New Methodologies and Compendial Perspective
Although the application of compendial methods is encouraged, there are many instances where the analytical method that best addresses the issue is a new method not found among the compendial methods. USP encourages the development of appropriate methods and submission of these methods, once validated, to the USP for addition to the compendia.
One such new methodology is the PCR-based assay. PCR-based assays are utilized in a variety of applications for both cell and gene therapy products. PCR assays can be a viable substitute for long-term bioassays and should be considered when a rapid-release strategy is used. Other applications include the use of PCR-based assays to assess adventitious viral agents in product or in cell and virus banks. PCR might also be used in an identity test of a gene therapy product. In all cases, validation of the new method and assay equipment and qualification of analytical personnel are required.
PCR assays are based on amplification of specific target DNA by using PCR technology. Traditionally, a pair of DNA oligonucleotide primers is used in combination with nucleotides and the Taq polymerase to increase the amount of a specific-size oligomer in a series of alternating cycles of permissive and nonpermissive polymerase temperature conditions. The reaction mix is resolved by gel electrophoresis and visualized by staining with ethidium bromide in order to detect the amplified target (amplicon). RT-PCR involves the use of a reverse transcriptase to create cDNA from RNA prior to performing the PCR reaction, so that the RNA can be detected. PCR and RT-PCR methods can be used qualitatively (positive or negative readout) or quantitatively.
Currently there are two common approaches to quantitation using PCR: (1) competitive PCR that involves use of a spiked mimic and (2) real-time or kinetic PCR that is based on the 5¢ nuclease activity of the Taq polymerase. In competitive PCR, quantitation is based on the ratio of the amplified mimic to the amplified target. In real-time PCR, the degradation of a dual-labeled, fluorescent, target-specific oligonucleotide probe is monitored in real time, as PCR amplification is occurring. The probe is labeled with a reporter fluorescent dye at the 5¢ end and a quencher fluorescent dye at the 3¢ end. When the probe is intact, the fluorescence emission of the reporter is quenched as a result of the physical proximity of the two dyes. The probe sequence is selected so that it hybridizes to the target between the two primer sites. During the extension phase of the PCR cycle, the probe is cleaved by the 5¢ nuclease activity of the Taq polymerase, while the reporter dye signal is increased by the release of reporter dye from the probe. Additional reporter dye molecules are cleaved during each cycle, resulting in an increase in the fluorescence intensity of the reporter dye proportional to the amount of amplicon produced. The resulting relative increase in reporter fluorescent dye emission is detected in real time during PCR amplification and it allows the threshold cycle number to be related to the target copy number. The threshold cycle number is defined as the PCR cycle number where the increase in reporter fluorescence is detected above the background fluorescence in the assay system. A greater quantity of input DNA or messenger RNA (mRNA) results in a lower threshold cycle, as a result of requiring fewer PCR cycles for reporter fluorescence-emission intensity to reach the threshold. Typically, assays can be designed to detect 1 to 10 copies of the target per reaction.
Like all analytical methods used to release product, PCR assays must be validated. Validation should include the rationale for the selection of primer and probe sequences and a demonstration of the specificity and efficiency of the primers and, for real-time quantitative PCR assays, of the probe. Because primers and probes are the main components of a nucleic acid–based detection system, the performance of the assay is highly dependent on the quality of these reagents. Specificity is generally demonstrated by assessing the resulting PCR product by gel electrophoresis to show that the amplicon is the expected size. For quantitative assays, the design and nature of the quantitation standards must also be addressed.
Assay validation must also address linearity, accuracy, ruggedness, and reproducibility with regard to both the assay itself and the sample preparation, that is, extraction of the sample DNA for PCR or of the sample RNA for RT-PCR. Validation should include a demonstration of the specific limit of detection in the sample type employed, because some sample types contain inhibitors of PCR. Validation should also address the reproducibility of the sampling scheme and the efficiency of nucleic acid extraction and purification procedures to produce the starting material (DNA or RNA). Well-designed spiking studies where samples are spiked both prior to and after extraction can address these issues.
PCR assays are occasionally subject to false-positive results because of the contamination of equipment or samples during handling in preparation for the assay. The most abundant source of contaminating target nucleic acid is the previously generated amplicon. However, the PCR reaction can be modified so that the resultant amplicon is sensitive to uracyl-N-glycosylase digestion and can therefore be eliminated. In addition, isolation of sample preparation areas from other phases of the assay and the use of dedicated equipment for each assay phase are generally necessary to prevent amplicon contamination of test samples and, hence, false-positive signals. Assay protocols that include appropriate controls, such as nontarget sequence and nucleic acid-free controls, can aid in determining the source and point of contamination if it occurs. Validation should address the procedures implemented to prevent contamination.
Sampling Issues
Sampling for lot-release testing should be based on the potential distribution for the parameter tested. See Stability-Protocol Development under Stability for additional considerations. Samples from each lot need to be retained in case there is a safety or quality issue with the lot. Even if the product has a very short shelf life, these retained samples can be used to detect impurities and other substances. The need for proper design of the sampling scheme is highlighted in safety testing for adventitious agents for cell or viral gene therapy products or in assessment of RCV for viral gene therapy products. In such cases, process validation will assist in determining the appropriate statistically based sampling design.
Safety
general considerations
Safety testing for cell and gene therapy products focuses on three issues: (1) preventing the unwitting use of contaminated cells, tissues, or gene therapy agents with the potential for transmitting infectious diseases, (2) preventing the use of improperly handled or processed, and consequently contaminated, products, and (3) ensuring safety when cellular and gene therapies are adapted for use other than in their normal functions or setting.
The primary means of assessing safety are the performance of biological assays to measure adventitious agents directly. Molecular biology-based assays that measure adventitious agent DNA or RNA are also used.
cell therapy products
Direct transmission of infectious disease is a major concern for cell therapy products. The degree of risk is dependent upon various factors such as whether the cells or tissues are to be used in a person different from the one they were obtained from; whether they are banked, shipped, or processed in a facility that handles cells and tissues from multiple donors; and how extensively they are processed. Improper handling can alter the integrity and function of cell therapy products by introducing microorganisms or by contaminating the therapeutic cell products with other donor or patient cells during collection, processing, or storage.
In addition to transmittable-disease screening and testing of allogeneic donors of all viable and nonviable tissues intended for use as cell therapy products, appropriate labeling and tracking are required. These requirements are not only based on the potential risk of disease transmission from donor to recipient but also on the following: (1) the unusual, but documented, possibility of product-to-product transmission (for example, viral contamination may occur among disrupted bags in the liquid phase of a liquid nitrogen freezer) or (2) the possibility of erroneous administration of a product to the wrong recipient. Specific donor screening and testing requirements for allogeneic cell products are based on those currently required for human blood products. These include (1) specific donor testing for HIV Type 1, HIV Type 2, hepatitis B, hepatitis C, and syphilis and (2) medical history screening for high risk for HIV, hepatitis B, Creutzfeldt-Jakob disease, and tuberculosis. Some of these tests and screening measures are also recommended, but not required, for autologous tissues.
The risk of cross-species infectivity during xenotransplantation is still unknown. Assessing the risk of infection from a new transmissible agent is difficult. In vitro coculture assays involving sensitive human indicator cell lines for the donor species should be developed. In particular, assay of endogenous retrovirus (ERV) present in the xenogeneic cell or tissue is required. In the case of porcine cells and tissues, both PCR and RT-PCR assays for porcine endogenous retrovirus (PERV) have been described and are applied to donor cells and tissues. These tests are also being used for patient monitoring. Assays for PERV antibody have also been developed for patient monitoring. Published studies indicate that the risk of PERV transmission to patients may be low.
Often the shelf life of cell therapy products is shorter than the time required to test for sterility and adventitious agents using traditional cell-based methods. However, as already discussed, development of validated rapid PCR-based methods allows both assessment and timely release. Presence of mycoplasma and a range of specific adventitious viruses and bacteria can be tested by using PCR or DNA- or RNA-hybridization dot blot analysis. Fourteen-day sterility testing is not always a viable alternative for final release of cell therapy products; in such cases, automated methods that rely on colorimetric detection or on continuous monitoring may be acceptable if they are validated. Facility and process validation are necessary adjuncts to ensure safety with regard to sterility and mycoplasma, particularly when rapid-release strategies are employed.
Additional testing for safety may be required when cell therapy products are used in the patient for a purpose other than that which the cells or tissue fulfills in its native state or when placed in a location of the body where such structural function does not normally occur. Testing should be designed to predict product behavior under these settings and should be designed based on the context of use. For example, in the case of a cell therapy product for cancer patients, where the cells are activated by culture on a feeder layer of cells during processing, it may be necessary to test the product cells for the presence of feeder cells. Residual feeder cells in the final product may cause an inflammatory response. In addition, products using non-human feeder cells are considered xenogeneic products. Cell therapies are exempted from general safety testing.
If the cells were modified by a viral gene vector during manufacturing, presence of RCV must be tested. Typically, RCV testing (see Viral Gene Therapy Products under Safety) is limited when rapid release is required by shelf life. Again, molecular biology–based methods such as PCR can be used in rapid screening situations. In such cases, during product development, testing that employs cell-based assays (for example, detection of cytopathic effect on indicator cell lines) is performed after release to validate the molecular biology–based test result.
viral gene therapy products
One of the primary safety concerns associated with viral vectors used for gene therapy is RCV. Regardless of the virus, these concerns are based on the potential lack of predictability for the pathogenicity of a contaminating virus for a specific route of administration, particularly if it is not the normal route of infection or if humans are not a natural host for the virus.
The pathogenesis of a wild-type adenovirus infection is known but may not be predictive for the routes of administration employed with recombinant adenoviral vectors. For adenoviral vectors, a limit of one RCA per dose is currently considered acceptable; other limits have been established for dose levels greater than 1 × 109 particles, specific indications, and routes of administration based on preclinical safety studies and patient-monitoring studies during clinical development. Limits as high as several thousand RCAs per dose have been reported. Typically, RCA levels are determined by using a cell-based assay that allows amplification of the RCA while preventing replication of the product. The cell line recommended for amplification and detection of RCA is the A549 cell line. However, some recombinant adenoviral vectors express therapeutic genes that interfere with analysis on A549 cells. In such cases, a bioassay utilizing two cell lines is used, with the first cell line chosen on the basis of resistance to the effects of expression of the therapeutic gene of interest and with subsequent passage of cell lysate onto A549 cells for amplification and detection of the RCA. RCA is most often detected by visual observation of the cytopathic effect but it may also be detected in the A549 cell culture by using immuno- or PCR-based methods. Quantitation of the RCA level is based on the quantity of sample tested and the detection limit of the assay. Typically, RCA bioassays are validated as being able to detect 1 plaque-forming unit or infectious unit of RCA in the test sample over a wide range of test-sample sizes. Test-sample sizes can range from 1 × 108 to 1 × 1012 particles but they are typically based on clinical-dose size. To verify detection limits, spike controls should be included as part of the test, even with validated assays. For recombinant adenoviruses produced using 293 cells, RCA detection by PCR methods can be confounded by detection of residual 293 host-cell DNA (detection of the E1 region). PCR assays, however, can be designed to specifically quantitate host cell DNA contamination and can be made specific to particular forms of slow-growing RCA. Quantitative PCR assays can be used in conjunction with a cell-based method for precise quantitation of RCA levels. When a tested sample is found to be positive, the identity of the RCA is usually confirmed by conducting PCR analysis. This rules out the possibility that contamination of the assay by exogenous wild-type adenovirus or other adventitious agents is responsible for the positive result.
For retroviral vectors, testing for RCR is required for cell banks, viral vector production lots, and any resultant ex vivo product lots (see the FDA's Draft Guidance for Industry: Supplemental Guidance on Testing for Replication Competent Retrovirus in Retroviral Vector Based Gene Therapy Products and During Follow-Up of Patients in Clinical Trials Using Retroviral Vectors, October 2000). Standard assays have been designed to detect replication-competent murine leukemia virus (MLV). The pathogenesis and potential long-term toxicity of low-level amphotropic MLV in human beings is not known. Methods commonly used to detect RCR include an amplification of virus titer by application of product to a replication-permissive cell line such as Mus dunni. Because infection is limited by the ability of a virus to reach the cells through Brownian motion, procedures (e.g., centrifugation and filtration) that physically bring the virus into contact with the cells may be used to enhance detection. However, high-titer recombinant vector can interfere with the detection of low-level RCR and this interference may be enhanced through such methods. Infected cells are passaged several times to allow viral replication. Culture medium is harvested at the end of the culture period and RCR detected by using an indicator cell line. If the product is an amphotropic MLV, RCR may be detected by using a feline cell-based PG4 S+L– assay, a mink cell-based MiCl S+L– assay, or a marker rescue assay. In S+L– assays, the RCR expresses proteins that lead to transformation and subsequent plaque formation on the monolayer. In a marker rescue assay, RCR infects a cell line that expresses a retroviral vector encoding a marker gene such as 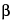 -galactosidase, drug resistance, or a fluorescent protein. The vector is packaged by the proteins supplied to it in trans by the RCR. The potentially vector-laden supernatant is transferred to naive target cells that are then screened for expression of the marker vector.
-galactosidase, drug resistance, or a fluorescent protein. The vector is packaged by the proteins supplied to it in trans by the RCR. The potentially vector-laden supernatant is transferred to naive target cells that are then screened for expression of the marker vector.
Testing for RCR is performed by cocultivation of the cell line or amplification of vector supernatant with an RCR replication-permissive cell line, typically Mus dunni, for several passages. Culture medium is harvested at the end of this cocultivation process and applied to an appropriate indicator cell line as described above. It is important to note that artifacts may be generated during the cocultivation assay by expression of an endogenous virus in the permissive cell line or through fusion if the vector-producing cell line is cultured directly with a marker rescue cell line. In addition, cocultivation may not be possible for ex vivo cell products that have specific culture requirements or limited culture life spans.
Methodologies for testing the presence of RCR in crude, purified bulk or final vector products are not specified. The FDA has deposited a reference standard of an amphotropic hybrid MLV with the ATCC. This viral stock has been assigned a label titer and should be used in assay validation. Method validation should demonstrate the ability to reproducibly detect a single RCR particle in individual product types because the product and its related impurities can interfere with the detection of RCR. Currently there are no acceptable limits for RCR contamination in products. Any product lot found to contain RCR cannot be used for human use.
Reference standards for assessing RCV in other viral vectors including ecotropic, xenotropic or pseudotyped MLV, adenovirus, and lentivirus have not been developed. Amplification and detection of replication-competent HIV, especially its pseudotyped variants, may warrant special containment and handling procedures.
Additional safety testing usually focuses on methods similar to those described under Biotechnology-Derived Articles 1045, in Safety Tests—Biologicals under Biological Reactivity Tests, In Vivo 88, and under Sterility Tests 71. For viral gene therapies produced using a human cell line, performance of the in vitro adventitious agent bioassays using 3 cell lines is recommended on either the bulk or final product. For adenoviral vectors, specific tests for adeno-associated virus are also recommended on either the bulk or final product. For adeno-associated virus, specific tests for adenovirus and herpesvirus are recommended on either the bulk or final product.
nonviral gene therapy products
Safety testing usually focuses on methods similar to those described under Biotechnology-Derived Articles 1045, in Safety Tests—Biologicals under Biological Reactivity Tests, In Vivo 88, and under Sterility Tests 71. However, the General Safety test is not required for therapeutic DNA plasmid products (even if formulated). Safety testing should be performed on nonviral formulated material. If the shelf life of the formulated nonviral therapy is very short, then the components should be tested individually.
Dose-Defining Assays
general considerations
An assay that precisely measures the amount of the product is referred to as a dose-defining assay, and it is selected on the basis of its precision and accuracy. An assay that measures therapeutic activity of the product is referred to as a potency assay and it is designed to measure product function. The design of the assay is dependent upon the type of product. In the case of drugs, the assays measuring the amount of active ingredient (dose) are referred to as strength assays.
Product dose can be defined as the concentration or amount of the drug product administered to the patient and it is typically measured as product mass. For cell and gene therapy products, attributes such as viable cell number, milligrams of plasmid or antisense oligonucleotide, or the viral particle number are often used to define the dose of the product.
cell therapy products
Cell therapy products may be dosed on the basis of enumeration of one or more cell populations. For ex vivo gene therapy, dose may be based on cell number as well as level of expression of the gene product. For products in the form of a homogeneous, single-cell suspension, viable cell number is the most frequently used assay. Such assays may include enumeration of all cells, total nucleated cells, or another subset of cells. Viability assays are usually based on a cell's ability to exclude a supravital dye, such as trypan blue. Results are expressed as the number of cells that exclude the dye and are therefore considered viable. The compound 7-AAD, a red-fluorescing compound that binds to nuclear proteins and is also excluded by viable cells, may be incorporated into flow-cytometric methods for simultaneous determination of viability and cell-identity markers.
Cell counting may be performed rapidly by manual or automated methods. Manual cell counting by visual enumeration of cells in a hemacytometer chamber is a readily available technique with acceptable accuracy, but a lower degree of precision than most automated methods. Typical instruments for automated cell counting provide reproducible enumeration of nonnucleated cells (e.g., erythrocytes and platelets) and nucleated cells and differential counting of the nucleated cells into mononuclear and polymorphonuclear leukocyte populations. Further discrimination of specific cell populations usually requires cell-surface phenotype analysis by flow-cytometric or other methods (see Cell Therapy Products under Identity). The proportion of a specific subpopulation of cells may be determined by FACS analysis or by flow cytometry.
An example of a cell enumeration assay is the enumeration of CD34-positive (CD34+) hematopoietic progenitor cells, the number being expressed as the number of cells per recipient's body weight. In numerous studies this measurement has been shown to predict hematologic reconstitution following myelosuppressive or ablative therapy in autologous or allogeneic hematopoietic transplantation.
For products that contain cells in a nonhomogeneous suspension, such as cells that form a two- or three-dimensional structure, alternative measures for cell enumeration, such as total area of a cell sheet, wet weight, total protein, and total DNA, have been used. If such measures are used to determine product dose, then supplemental tests must be performed to demonstrate therapeutic activity.
viral and nonviral gene therapy products
Particle concentration is a commonly used measure for viral vector product dose. Particle concentration may be measured by physical, biophysical, or in vitro cell-based assays. For example, quantitation of purified adenoviral particles may be determined by using the optical density of a solution of virus in 0.1% (w/v) sodium dodecyl sulfate (SDS) solution, at 260 nm, because a relationship between absorption and particle concentration has been published for adenovirus. The particle number concentration is equivalent to the product of the absorbance at 260 nm in a 1-cm cell, the dilution factor, and 1.1 × 1012 particles.2 Other methods to determine particle concentration include particle counting by electron microscopy and integration of viral peak area against an authenticated reference standard in an anion-exchange resin-based high-pressure liquid chromatographic (HPLC) assay.
Virus concentration can also be assessed through the measurement of selected structural proteins with known molecular masses and known copy numbers within the virion. For this method, the virus has to be lysed, and the structural proteins have to be separated by using an appropriate, high-recovery chromatographic procedure (e.g., reverse-phase HPLC). The chromatographic separation and the identity and the purity of the selected structural protein has to be verified during assay validation by methods such as SDS polyacrylamide gel electrophoresis (SDS-PAGE), peptide sequencing, and mass spectroscopy. The selected structural proteins have to be quantified, for example, by integrating chromatographic peaks at 214 nm and comparing the area to an authenticated reference standard. The virus concentration can then be calculated based on the molecular mass, the copy number, and the measured mass of the protein. Very importantly, the virus concentration can be estimated simultaneously for multiple structural proteins, allowing the use of this assay in relatively impure virus preparations. This method has been applied to adenovirus and should be applicable to other viral vector types.
Biophysical methods of determining particle number include direct quantitation of vector nucleic acid by radiolabeled-probe hybridization and indirect quantitation by amplification of template nucleic acid (e.g., PCR and RT-PCR) or by signal amplification (e.g., branched-chain DNA using multiple-probe hybridization).
In cases where biophysical methods are not available, bioassays that measure gene-vector titer have been used. These involve infection, transfection, or transduction of a susceptible cell line in vitro, followed by some measure of the product uptake. Methods for quantitation or estimation of the number of infection, transfection, or transduction events include plaque-forming unit assays, tissue culture infectious dose, 50% (TCID50) assays based on cytopathic effect or immunofluorescent detection of an expressed vector protein, or a quantitative DNA-hybridization assay. Examples are as follows.
For retroviral or lentiviral gene therapy products or AAVs that carry a selectable marker (e.g., that for neomycin resistance) or a reporter gene (e.g., -galactosidase) in addition to the therapeutic gene, the infectious titer is commonly determined by measuring the number of transduced or infected cells expressing these nontherapeutic proteins. Vector titer is typically reported as the number of colony-forming units (cfu) per mL for cells transduced with viral vectors containing drug-resistance markers and selected for growth in drug-containing medium. Titer based on -galactosidase can be expressed in terms of blue (cfu) per mL after staining and counting the cells that convert the -galactosidase substrate X-Gal into a blue chromophore. For vectors without a marker gene, quantitation of transduction has been measured precisely by using quantitative PCR.
Most nonviral gene therapy products contain plasmid DNA and their usual measure of dose is the DNA mass. The DNA mass may be determined in the formulated state, and, if recombinant protein is included in the formulation, the total combined mass of all formulation components based on a specific ratio can be used. DNA concentrations greater than 500 ng per mL are most simply determined by using optical density measurement at 260 nm. This method is not generally applicable to lipid-formulated DNA. Because RNA and proteins also have significant absorbances at 260 nm, other analyses must be performed to demonstrate that there is minimal contamination with RNA, protein, or residual host-cell chromosomal DNA. Dyes that specifically bind to double-stranded DNA allow the DNA concentrations of less than 500 ng per mL to be measured accurately when calculated against an authenticated DNA standard curve. PicoGreen is one such fluorescent dye and it is minimally affected by single-stranded DNA, RNA, proteins, salts, and detergents. The fluorescent dye Hoechst 33258 also binds to both double-stranded and single-stranded DNA and it can be used to determine DNA concentrations as low as 0.3 ng per mL. The Hoechst 33258 does not bind to protein or RNA and it can accurately determine the DNA concentrations in crude samples.
Methods, such as capillary electrophoresis, employing an authenticated reference material, can also be used to determine the strength of nonviral and antisense-oligonucleotide products.
Potency
general considerations
Potency is defined as the therapeutic activity of the drug product. Together with dose, potency defines the biological activity of each lot (see General Considerations under Dose-Defining Assays). Potency may be assessed through in vitro or in vivo bioassays. It is not uncommon for these assays to have coefficients of variation between 30% and 50%. These assays require a well-defined, representative reference material that can be used as a positive control for the assay. The positive control serves to qualify the performance of an individual assay. Potency assay development should focus on characterizing and controlling variability. The high-precision assays are more effective tools in monitoring product quality. Information about potency-assay variability should be incorporated into the stability study design and the proposed statistical approach to assignment of expiration date (see Stability).
cell therapy products
Functional assays that can be performed on cellular products are application-related and include viable cell number and a wide range of colony-forming assays, proliferative assays, cell-to-target killing assays, and assays that quantitate gene expression following gene transduction. For hematopoietic progenitor cells prepared from marrow, peripheral blood, or cord blood, traditional colony-forming assay quantitates committed progenitor cells such as colony-forming unit-granulocyte–macrophage (CFU-GM); this assay has been correlated with clinical engraftment outcomes in some studies. More recently, process-monitoring programs incorporate assays such as the long-term culture-initiating cell (LTCIC) assay or the in vivo animal models such as competitive repopulation in immunodeficient mice to monitor the activity of the most primitive hematopoietic stem cells. In the case of cells intended for structural repair, proliferation under a set of defined ex vivo conditions may be used as the potency assay. If the cells release an enzyme or active molecule, a potency assay could be based on the units of enzymatic activity or on the total of active molecules released. For instance, the production of insulin in response to changes in glucose levels could be the basis of a potency assay for cells intended to treat diabetes.
Patient-specific products, such as autologous cancer vaccines that elicit an in vivo immune response, present a challenge in demonstrating therapeutic activity in an in vitro or in vivo assay system. Assessment of potency in these circumstances is currently the subject of public-policy debate. Novel approaches to measuring potency, such as the correlation of clinical outcome to other characterization tests such as identity tests, may be appropriate and should be discussed with regulatory authorities early in development. For example, the ability to determine specific cell-surface identity markers by employing flow-cytometric techniques or vital stains may be an acceptable measurement of potency if properly validated and correlated with clinical outcome.
viral and nonviral gene therapy products
Bioassays employed to measure the potency of viral and nonviral gene therapy products generally involve infection, transfection, or transduction of a susceptible cell line in vitro, followed by some functional measure of the expressed gene of interest. Functional assays for the therapeutic gene (e.g., those measuring enzyme activity and cytokine activity) should generally be used instead of analytical methods such as enzyme-linked immunosorbent assay (ELISA), HPLC, or FACS, which provide information about the level of expression but only infer function. In addition, for viral vectors, infectious titer measurements by themselves are generally not considered an adequate measure of product potency. The design and ultimate suitability of an assay system for determining product potency will depend on the relationship between the intended human target cell in vivo and the following: (1) the transduction or transfection efficiency of the cell line used in vitro (2) the protein expression levels, and (3) the duration of expression required for the therapeutic effect.
In vivo tests may also be used to measure vector-product potency. Readouts may be based on a response per animal (e.g., blood levels of therapeutic protein 24 hours after treatment) or a group response rate (e.g., percentage of animals that elicited an immune response or survived virus challenge). The availability of an appropriate in vivo test system will depend on the vector-host range (for viral vectors), the pharmacokinetics and biodistribution of the vector and its resultant gene product relative to its human counterpart, and the time frame required to observe the therapeutic effect or surrogate. Issues of cost, facilities, validation, and ethics will determine the practicality of an in vivo potency test.
Purity
general considerations
Analytical methods that separate, isolate, and specifically quantify the intended active product components determine product purity. Impurities are either product- or process-related components that can be carried through to the final product. The manufacturing and purification process should be optimized to consistently remove impurities while retaining product activity. The requirement to test for a particular impurity for product lot release will depend on the following: (1) the demonstrated capability of the manufacture and purification process to remove or inactivate the impurity through process validation, and (2) the toxicity potential associated with the impurity.
Examples of process-related impurities associated with cell and gene therapy products include residual production-medium components (e.g., FBS, antibiotics, cytokines, and Escherichia coli chromosomal DNA in a plasmid product), ancillary products used in downstream processing (e.g., nucleases such as DNase I), and residual moisture for lyophilized vector products. Impurities may be bioactive (e.g., cytokines and hormones) or immunogenic (e.g., product aggregates, degradation products, plasmid-selection markers, and nonhuman-derived proteins) or they may have other deleterious effects (e.g., they may compete with the product) if administered at a dose equal to that of the product. Product-related impurities are specific to each product type. Examples include differentiated cells in a stem-cell therapy product, nicked plasmid forms in nonviral products, and defective or immature virus particles in retroviral or adenoviral vector products. Analytical methodologies to assess purity require quantitation or physical separation of intended product from its impurities. Common sense should drive the need to quantify specific impurities. It may be possible to validate the manufacturing process to the extent that specific lot-release testing for impurities will be very limited. An emphasis may be placed on demonstrating the consistency of the product-impurity profile.
Testing for impurities is often extensive during product characterization and process validation when the consistency of the manufacturing and purification process is being demonstrated. Testing for impurities as part of lot-release testing should reflect the safety risks associated with the impurity and the ability of the process to consistently remove that impurity.
cell therapy products
One measure of purity is the percentage of viable cells in the total cell population. Another measure is the percentage of transduced cells or the percentage of cells with a specific marker. If an entire population of cells is the therapeutic agent, methods should determine the relative amounts of each subpopulation. Limits should be defined for each cell subpopulation. These assays may be based on immunological methods utilizing flow cytometry or DNA-hybridization dot blot analysis. Additional tests for process contaminants are performed depending on the specifics of the manufacturing process. For example, a quantitative ELISA for residual serum proteins may be required. If the cells are genetically modified during manufacture, then testing for residual vector may need to be performed.
For gene-modified cell therapies, determining the purity of the cell therapy product depends on the availability of reagents and methods to distinguish therapeutic cells from the other cells present in the product. As in the case of gene therapy products, gene-modified cells can be distinguished from the unmodified cells on the basis of expression of the transgene. FACS using an antibody that detects the therapeutic protein or fluorescent probes that detect expressed RNA allow the separation and quantitation of the transgene expressing and nonexpressing cells. By adding an antibody to a cell-specific phenotypic marker and by using a double-sorting technique, FACS can be used to further identify the subpopulation of cells that are modified.
Endotoxin testing is also required. Biomaterials used with cell therapy products should also be tested for their biocompatibility.
viral gene therapy products
Product-related impurities for viral vectors include aggregates and defective and immature particles that may be produced during the manufacture or purification of the recombinant vector. Aggregates of vector may form if the product is highly concentrated, stored under certain conditions (e.g., under certain pH or temperature), or reconstituted after lyophilization. Assays to detect aggregates include particle size analysis by laser light-scattering and the use of nonreducing, nondenaturing PAGE, followed by staining of the gel or transfer and detection of viral proteins by Western blot analysis. Sedimentation rate analysis also allows separation of aggregates from monomers based on size. Optical density analyses of light-scattering are also used to assess vector aggregation.
Defective particles are viral particles that do not contain the appropriate recombinant genome, that is, they contain some other nucleic acid or contain no genome at all, or the vector has some missing, defective, or otherwise altered structural component that impairs its ability to transduce a cell. For viral vector systems that have capsomeric symmetry, which requires the appropriate nucleic acid incorporation for configuration, empty particles may be readily distinguished from those carrying genomes. For enveloped viruses, empty particles may not be as readily separated from those with encapsidated nucleic acid.
For some viral vector products, active viral particles may be separated from defective particles by using analytical HPLC. Anion-exchange resins have been used to separate active adenovirus from defective virus particles. However, this method might not be useful for an adenoviral vector purified by anion-exchange chromatography unless the resin for the assay is different from that used during manufacture. Depending on the nature of the viral vector and its nonactive or defective forms, other methods of separation, such as equilibrium centrifugation in a cesium chloride density gradient, may need to precede the quantitation of the active particle. Ideally, the method of separation will allow quantitation.
Defective particles that carry a noncell-derived oncogenic gene or other undesirable genes may pose a special concern. For example, in murine-based retroviral packaging cell lines, small viral elements called VL30 sequences can be packaged in about one third of all particles. Assays may need to be developed to quantify specific defective particles if they are known to be present in quantities sufficient to pose a safety concern.
Virus quality and the comparability of preparations can also be assessed by measuring selected structural proteins with known molecular masses and known copy numbers within the virion. For this method, the virus is lysed, and the structural proteins are separated by using reverse-phase HPLC or some other high-recovery chromatographic procedure. The chromatographic separation should be validated and the identity of the selected structural proteins verified by methods such as SDS-PAGE, peptide sequencing, or mass spectroscopy. One can fingerprint the batch based on quantification of the selected structural proteins and comparison to a reference standard. When the method incorporates the use of mass spectroscopy, impurities such as structural variants can also be identified. For adenovirus preparations, some precursor and most mature virion proteins can be monitored, thus allowing monitoring of the product and of the immature virion forms.
Host cell-derived proteins may be considered impurities for some viral vector products and may be separated and quantified by PAGE or HPLC or detected by amino acid analysis, Western blot, or immunoassay-based methods. However, for enveloped viruses such as retroviruses, host cell-derived membrane proteins are an integral part of the product. In those vector systems, it may be difficult to determine the presence of contaminating exogenous host-derived proteins.
Presence of specific process-related impurities depends on the manufacture and purification process of each vector or product type. However, most products will need to be tested for residual endotoxin (see Bacterial Endotoxins Test 85). Acceptable limits of endotoxins have been determined and can be directly applied to viral vector products.
Although genomic DNA derived from continuous cell substrates used to manufacture biological product has been considered historically as potentially tumorigenic, recent studies suggest that the risks are very low. However, every attempt should be made during process development to reduce contaminating DNA levels. The need to test for residual DNA as part of product lot release should be evaluated on a case-by-case basis and may be dependent upon the size distribution of the DNA, its association with the product or its formulation components, and the route of administration of the product. Quantitative PCR assays have been developed to analyze the amount of residual host-cell DNA by using primers designed to amplify evolutionarily conserved and abundant target sequences, such as 18S for 293 cells.
Quantitation of residual serum components such as bovine serum albumin (BSA) can be achieved by using ELISA and a BSA reference standard. Specific functional or immunological methods may need to be developed for other ancillary products including other culture media or purification process components such as cytokines or enzymes (e.g., nucleases such as DNase I or benzon nuclease).
nonviral gene therapy products
Testing is usually performed on the individual components, the plasmid DNA, the lipid or lipoplex components and (recombinant) protein components if any are present in the formulation. Plasmid DNA is characterized for a variety of impurities including residual host-cell DNA, residual RNA, and residual protein. Residual protein testing is frequently included in lot-release testing. Optical density ratios, usually the ratio of the measurement at 260 nm to that at 280 nm, are frequently used in purity specifications for plasmid DNA.
In addition, the plasmid DNA should also be characterized with regard to its form. Plasmid DNA forms include monomeric supercoiled, relaxed monomer, and linear forms. The profile of forms needs to be monitored for product consistency. Additionally, it may be possible to correlate form with in vivo transfection behavior. While monomeric supercoiled plasmid has been shown to be more efficient than relaxed monomer, linear, or multimeric forms of the plasmid in transfecting cell lines in vitro, in vitro transfection has been shown to not always predict in vivo behavior. Formulation, delivery method, and route may impact in vivo transfection. Agarose gel electrophoresis can resolve these forms of plasmid, which are then detected by UV after ethidium bromide staining. This method provides information about the relative levels of the plasmid forms, but it is not highly quantitative for the individual species. Analytical anion-exchange HPLC can be used as a quantitative assay for monomeric supercoil and percentage of other forms, including concatamers. Other sophisticated methods, such as capillary zone electrophoresis, linear-flow dichroism, and atomic-force microscopy have been proposed as replacements for agarose gel analysis. Until they are validated, these analytical methods may be more appropriate for characterization studies in support of process development and validation rather than for lot-release testing. The appropriate methods for lot release will depend on what effect these alternate plasmid forms have on the product potency. Many of these methods, such as HPLC, are also applicable to the assessment of the purity of antisense-oligonucleotide products and the determination of the level of by-products.
Tests for process-related impurities, such as cesium chloride, must also be conducted. In the case of antisense-oligonucleotide products, residual solvents must be quantified. Lipid and lipoplex formulation components must also be tested for their chemical purity. Testing for specific chemical impurities is commonly performed by using gas chromatography–mass spectroscopy (GC–MS), high-pressure liquid chromatography (HPLC), or thin-layer chromatography (TLC) methods.
If protein is part of the formulated complex, then the protein must also be tested for purity. The methods outlined under Biotechnology-Derived Articles 1045 or under Biotechnology-Derived Articles—Capillary Electrophoresis 1053, Biotechnology-Derived Articles—Isoelectric Focusing 1054, and Biotechnology-Derived Articles—Polyacrylamide Gel Electrophoresis 1056 are relevant.
Bacterial protein, DNA, RNA, and endotoxins are the major types of host-derived process contaminants. Standard protein assays (e.g., Lowry, Bradford, or Coomassie), PAGE followed by silver staining or Western blot analysis, or ELISA can be used to detect residual host protein in the nanogram range. Host chromosomal DNA may be detected by slot blot hybridization (detection in picogram range) or by PCR using highly conserved target sequences (e.g., 18S for Escherichia coli). However, low background may be unavoidable in PCR-based assays because the recombinant polymerases used for the amplification of target also contain residual bacterial DNA. PAGE or agarose gel electrophoresis followed by fluorescent dye staining may be used to detect residual RNA. Quantitation may not be required given the labile nature of RNA and the low-level toxicity associated with it.
Certain antibiotics, such as kanamycin, that may be used during the fermentation process must be removed during the process, and validation of the process or lot-release testing must be performed to confirm removal during the purification of the plasmid. HPLC is one method that can be used to detect low-level residual antibiotic.
lyophilized viral and nonviral vector products
Residual moisture may affect the stability of a lyophilized vector product. The FDA's Guideline for the Determination of Residual Moisture in Dry Biological Products (January 1990) recommends a 1% residual moisture level, although data indicating no adverse effects on product stability at higher levels will be considered acceptable. Residual moisture levels can be determined by using a standard method (see Water Determination 921) that is compatible with the formulated product.
Identity
general considerations
Lot-release testing for cell and gene therapy products must include an identity test. This test serves to specifically identify the product. The complexity of the identity test will depend on the nature of the specific product and the array of products being manufactured. For example, more extensive and rigorous testing may be performed for an autologous gene-modified cell therapy product at a facility where multiple patient products are manufactured than for a viral vector product produced at a site that manufactures a single vector product.
cell therapy products
Cell therapy identity tests must be relevant to the cell type and manipulations applied during processing. Differential surface markers (for instance, CD3, CD4, CD34, and CD45) are frequently used to ascertain product identity. Flow-cytometric immunoassay methods are the most common means of detecting and quantifying these markers. In this type of assay, a sample of the cells is stained with fluorescently labeled antibodies directed against specific identity markers and then passed as a single cell suspension in front of a laser source. Identification and quantitation of particular cell subsets is accomplished by multiparameter analysis, usually of size and granularity (measured by forward and side light-scattering) and of one or more identity markers (measured by emitted fluorescence). Simultaneous quantitation of cell viability can be performed by adding 7-amino-actinomycin D (7-AAD) to cell suspensions marked with antibodies conjugated to green (e.g., FITC) or orange (e.g., phycoerythrin) fluorescent compounds.
Analyses, such as isoenzyme analyses, employing biochemical markers are also used. For example, isoenzyme analyses are used to confirm species in the case of xenotransplants. Cell morphology can be used if it can distinguish specific cell types or unique function. Morphology can be combined with doubling-time parameters to better distinguish different cell types.
viral gene therapy products
Restriction enzyme mapping and sequencing of the transcription unit DNA are the most commonly used approaches to establishing the identity of viral vectors for characterization purposes. PCR-based methods, restriction enzyme mapping, and transgene expression–based immunoassays are most commonly used to confirm the identity during lot-release testing.
nonviral gene therapy products
Restriction enzyme mapping is the most common identity method for plasmid-DNA and antisense-oligonucleotide products. The number of enzymes used to create the vector fingerprint will vary with the complexity of the DNA and the degree of similarity between multiple products. If lipids, lipoplex agents, or proteins are used to formulate the DNA, then their identity must also be tested. Lipids and lipoplex chemicals may be identified by procedures used for traditional pharmaceuticals, such as GC–MS, TLC, and the like. Protein components of the formulation may be identified by peptide mapping or other means outlined under Biotechnology-Derived Articles—Isoelectric Focusing 1054, Biotechnology-Derived Articles—Peptide Mapping 1055, and Biotechnology-Derived Articles—Polyacrylamide Gel Electrophoresis 1056.