- British Pharmacopoeia Volume I & II
- Monographs: Medicinal and Pharmaceutical Substances
Bupivacaine Hydrochloride |
 |


(Ph. Eur. monograph 0541)
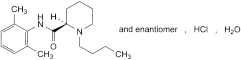
C18H28N2O,HCl,H2O 342.9 73360-54-0
Local anaesthetic.
Bupivacaine and Adrenaline Injection/Bupivacaine and Epinephrine Injection
Bupivacaine and Diamorphine Injection
Bupivacaine and Fentanyl Injection
Ph Eur
(2RS)-1-Butyl-N-(2,6-dimethylphenyl)piperidine-2-carboxamide hydrochloride monohydrate.
98.5 per cent to 101.0 per cent (dried substance).
White or almost white, crystalline powder or colourless crystals.
Soluble in water, freely soluble in ethanol (96 per cent).
First identification A, D, E.
Second identification B, C, D, E.
A. Infrared absorption spectrophotometry (2.2.24).
Comparison bupivacaine hydrochloride CRS.
B. Thin-layer chromatography (2.2.27).
Test solution Dissolve 25 mg of the substance to be examined in methanol R and dilute to 5 mL with the same solvent.
Reference solution Dissolve 25 mg of bupivacaine hydrochloride CRS in methanol R and dilute to 5 mL with the same solvent.
Plate TLC silica gel G plate R.
Mobile phase concentrated ammonia R, methanol R (0.1:100 V/V).
Application 5 µL.
Development Over a path of 10 cm.
Drying In air.
Detection Spray with dilute potassium iodobismuthate solution R.
Results The principal spot in the chromatogram obtained with the test solution is similar in position, colour and size to the principal spot in the chromatogram obtained with the reference solution.
C. Dissolve 0.1 g in 10 mL of water R, add 2 mL of dilute sodium hydroxide solution R and shake with 2 quantities, each of 15 mL, of 1,1-dimethylethyl methyl ether R. Dry the combined upper layers over anhydrous sodium sulfate R and filter. Evaporate the filtrate, recrystallise the residue from ethanol (90 per cent V/V) R and dry under reduced pressure. The crystals melt (2.2.14) at 105 °C to 108 °C.
D. It gives reaction (a) of chlorides (2.3.1).
E. Optical rotation (see Tests).
Dissolve 1.0 g in carbon dioxide-free water R and dilute to 50 mL with the same solvent.
Solution S is clear (2.2.1) and colourless (2.2.2, Method II).
To 10 mL of solution S add 0.2 mL of 0.01 M sodium hydroxide; the pH (2.2.3) is not less than 4.7. Add 0.4 mL of 0.01 M hydrochloric acid; the pH is not greater than 4.7.
-0.10° to + 0.10°.
Dissolve 1.0 g in methanol R and dilute to 20.0 mL with the same solvent.
Gas chromatography (2.2.28).
Internal standard solution Dissolve 25 mg of methyl behenate R in methylene chloride R and dilute to 500 mL with the same solvent.
Test solution Dissolve 50.0 mg of the substance to be examined in 2.5 mL of water R, add 2.5 mL of dilute sodium hydroxide solution R and extract with 2 quantities, each of 5 mL, of the internal standard solution. Filter the lower layer.
Reference solution (a) Dissolve 10 mg of the substance to be examined, 10 mg of bupivacaine impurity B CRS and 10 mg of bupivacaine impurity E CRS in 2.5 mL of water R, add 2.5 mL of dilute sodium hydroxide solution R and extract with 2 quantities, each of 5 mL, of the internal standard solution. Filter the lower layer and dilute to 20 mL with the internal standard solution.
Reference solution (b) Dilute 1.0 mL of the test solution to 100.0 mL with the internal standard solution.
Reference solution (c) Dilute 5.0 mL of reference solution (b) to 10.0 mL with the internal standard solution.
Reference solution (d) Dilute 1.0 mL of reference solution (b) to 10.0 mL with the internal standard solution.
- — material: fused silica;
- — size: l = 30 m, Ø = 0.32 mm;
- — stationary phase: poly(dimethyl)(diphenyl)siloxane R (film thickness 0.25 µm).
Carrier gas helium for chromatography R.
Flow rate 2.5 mL/min.
Split ratio 1:12.
Temperature:

Detection Flame ionisation.
Injection 1 µL.
Identification of impurities Use the chromatogram obtained with reference solution (a) to identify the peaks due to impurities B and E.
Relative retention With reference to bupivacaine (retention time = about 10 min): impurity B = about 0.7; impurity E = about 1.1; internal standard = about 1.4.
System suitability Reference solution (a):
- — resolution: minimum 3.0 between the peaks due to bupivacaine and impurity E.
- — impurity B: calculate the ratio (R1) of the area of the principal peak to the area of the peak due to the internal standard from the chromatogram obtained with reference solution (c); from the chromatogram obtained with the test solution, calculate the ratio of the area of the peak due to impurity B to the area of the peak due to the internal standard: this ratio is not greater than R1 (0.5 per cent);
- — unspecified impurities: calculate the ratio (R2) of the area of the principal peak to the area of the peak due to the internal standard from the chromatogram obtained with reference solution (d); from the chromatogram obtained with the test solution, calculate for each impurity the ratio of the area of any peak, apart from the principal peak, the peak due to impurity B and the peak due to the internal standard, to the area of the peak due to the internal standard: this ratio is not greater than R2 (0.10 per cent);
- — total: calculate the ratio (R3) of the area of the principal peak to the area of the peak due to the internal standard from the chromatogram obtained with reference solution (b); from the chromatogram obtained with the test solution, calculate the ratio of the sum of the areas of any peaks, apart from the principal peak and the peak due to the internal standard, to the area of the peak due to the internal standard: this ratio is not greater than R3 (1.0 per cent);
- — disregard limit: ratio less than 0.05 times R3 (0.05 per cent).
Liquid chromatography (2.2.29). Prepare the solutions immediately before use.
Test solution Dissolve 50 mg of the substance to be examined in mobile phase A and dilute to 10.0 mL with mobile phase A.
Reference solution (a) Dissolve 5.0 mg of bupivacaine impurity F CRS in mobile phase A and dilute to 100.0 mL with mobile phase A. Dilute 1.0 mL of the solution to 100.0 mL with mobile phase A. Dilute 1.0 mL of this solution to 10.0 mL with mobile phase A.
Reference solution (b) Dissolve 20 mg of methyl benzoate R and 25 mg of bupivacaine impurity F CRS in mobile phase A and dilute to 50.0 mL with mobile phase A. Dilute 3.0 mL of the solution to 50.0 mL with mobile phase A. Dilute 1.0 mL of this solution to 10.0 mL with mobile phase A.
- — size: l = 0.25 m, Ø = 4.6 mm;
- — stationary phase: end-capped octadecylsilyl silica gel for chromatography R (5 µm).
- — mobile phase A: dissolve 0.23 g of sodium dihydrogen phosphate monohydrate R and 3.626 g of disodium hydrogen phosphate dihydrate R in water R and dilute to 1000 mL with the same solvent; mix equal volumes of this solution (pH 8.0) and acetonitrile R;
- — mobile phase B: acetonitrile R;

Flow rate 1.0 mL/min.
Detection Spectrophotometer at 240 nm.
Injection 50 µL.
Identification of impurities Use the chromatogram obtained with reference solution (a) to identify the peak due to impurity F.
Relative retention With reference to bupivacaine (retention time = about 20 min): impurity F = about 0.3; methyl benzoate = about 0.4.
- — resolution: minimum 4.0 between the peaks due to impurity F and methyl benzoate in the chromatogram obtained with reference solution (b);
- — signal-to-noise ratio: minimum 40 for the principal peak in the chromatogram obtained with reference solution (a).
- — impurity F: not more than the area of the principal peak in the chromatogram obtained with reference solution (a) (10 ppm).
Maximum 10 ppm.
Dissolve 2.0 g in a mixture of 15 volumes of water R and 85 volumes of methanol R and dilute to 20 mL with the same mixture of solvents. 12 mL of the solution complies with test B. Prepare the reference solution using lead standard solution (1 ppm Pb) obtained by diluting lead standard solution (100 ppm Pb) R with a mixture of 15 volumes of water R and 85 volumes of methanol R.
4.5 per cent to 6.0 per cent, determined on 1.000 g by drying in an oven at 105 °C.
Maximum 0.1 per cent, determined on 1.0 g.
Dissolve 0.250 g in a mixture of 20 mL of water R and 25 mL of ethanol (96 per cent) R. Add 5.0 mL of 0.01 M hydrochloric acid. Carry out a potentiometric titration (2.2.20), using 0.1 M ethanolic sodium hydroxide. Read the volume added between the 2 points of inflexion.
1 mL of 0.1 M ethanolic sodium hydroxide is equivalent to 32.49 mg of C18H29ClN2O.
Protected from light.
Specified impurities B, F.
Other detectable impurities (the following substances would, if present at a sufficient level, be detected by one or other of the tests in the monograph. They are limited by the general acceptance criterion for other/unspecified impurities and/or by the general monograph Substances for pharmaceutical use (2034). It is therefore not necessary to identify these impurities for demonstration of compliance. See also 5.10. Control of impurities in substances for pharmaceutical use): A, C, D, E.
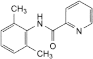
A. N-(2,6-dimethylphenyl)pyridine-2-carboxamide,
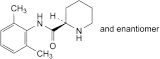
B. (2RS)-N-(2,6-dimethylphenyl)piperidine-2-carboxamide,
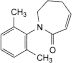
C. 1-(2,6-dimethylphenyl)-1,5,6,7-tetrahydro-2H-azepin-2-one,
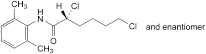
D. (2RS)-2,6-dichloro-N-(2,6-dimethylphenyl)hexanamide,
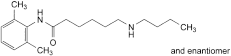
E. 6-(butylamino)-N-(2,6-dimethylphenyl)hexanamide,
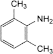
F. 2,6-dimethylaniline.
Ph Eur