BIOWAIVER
The term biowaiver is applied to a regulatory approval process when the application (dossier) is approved on the basis of evidence of equivalence other than an in vivo BE test. For solid oral dosage forms, the evidence of equivalence is determined on the basis of an in vitro dissolution profile comparison between the multisource and the comparator product.
Biowaiver Based on the Pharmaceutical Dosage Form
A drug product's in vivo comparative BA or BE study requirement may be waived if the products compared contain the same API(s) in the same concentration, contain the same excipients in comparable concentrations, and meet one of the following criteria:
- Aqueous solutions to be administered parenterally
- Solutions for oral use that do not contain an excipient that is known or is suspected to affect gastro-intestinal transit or absorption of the active substance
- Gases
- Powders for reconstitution as a solution
- Otic or ophthalmic products prepared as aqueous solutions
- Topical products prepared as aqueous solutions
- Inhalation products or nasal sprays tested to be administered with essentially the same device. Special in vitro performance testing should be required to document comparable device performance.
Biowaiver Based on Dosage Form Proportionality
When a single-dose fasting BE study is conducted on the designated (usually highest) strength of the drug product, the requirement for the conduct of additional in vivo BE studies on the lower strengths of the same product can be waived, provided that the lower strength (1) is in the same dosage form; (2) is proportionally similar in its active and inactive ingredients; (3) has the same drug release mechanism (for extended-release products); (4) meets an appropriate in vitro dissolution profile comparison criterion (f2 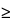 50); and (5) both lower and higher strengths are within the linear pharmacokinetic range.
50); and (5) both lower and higher strengths are within the linear pharmacokinetic range.
Biowaiver Based on the Biopharmaceutics Classification System
The BCS is based on aqueous solubility and intestinal permeability of the API. When the properties of the API are evaluated in conjunction with the dissolution of the pharmaceutical dosage form, the BCS takes into account three major factors that govern the rate and extent of drug absorption from immediate-release dosage forms. On the basis of the solubility and permeability of the dosage form, the drug substance is placed in one of four classes:
Class 1: high solubility, high permeability
Class 2: low solubility, high permeability
Class 3: high solubility, low permeability
Class 4: low solubility, low permeability
Use of the BCS has become a means of documenting BE without the conduct of an in vivo study; see the FDA Guidance Guidance for Industry: Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System (2000) (http://www.fda.gov/; search by document title).
The in vitro dissolution studies are generally carried out by basket method at 100 rpm or by paddle method at 50 rpm (FDA Guidance cited immediately above) or 75 rpm [WHO Guidance, Annex 7 Multisource (Generic) Pharmaceutical Products: Guidelines on Registration Requirements to Establish Interchangeability (2006)] http://www.who.int/en/; search by document title) in 900 mL of medium at pH 1.2, 4.5, and 6.8. On the basis of dissolution rate, the pharmaceutical dosage forms are classified as (1) very rapidly dissolving, if 85% or greater of the the dosage form dissolves in 15 minutes or less; (2) rapidly dissolving, if 85% or greater of the dosage form dissolves in 30 minutes; or (3) slowly dissolving, if the dosage form takes more than 30 minutes for 85% of drug dissolution.
For biowaiver, the dissolution tests should be carried out for both generic and reference product under the same test conditions. For the generic product to be eligible for biowaiver, the reference product should belong to the same BCS class and should meet dissolution profile comparison criteria. Based on BCS classification and dissolution profile comparison, biowaiver can be considered by regulatory authorities provided the dissolution profile similarity criteria provided in the next sections are met.
Class 1 Drug Products (Allowed in WHO and FDA Approaches)
Dosage forms of drug substances that are highly soluble, highly permeable, and rapidly dissolving are eligible for biowaivers under the following conditions:
-
85% or more of the dosage form dissolves in 30 minutes or less and the dissolution profile of the generic product is similar to that of the reference product in pH 1.2, 4.5, and 6.8 buffer, using the basket method at 100 rpm or the paddle method at 50 rpm (FDA) or 75 rpm (WHO), and meets the criterion of dissolution profile similarity, f2 50.
- If both the reference and the generic dosage forms are very rapidly dissolving (i.e., 85% dissolution in 15 minutes or less in all three media under the above test conditions), then profile determination is not necessary.
Class 2 Drug Products (WHO Approach)
Dosage forms of drug substances with high solubility only in pH 6.8 and high permeability (low solubility by definition, BCS Class 2) are eligible for biowaivers, provided that:
- The dosage form is rapidly dissolving (85% or more in 30 minutes or less) in pH 6.8 buffer.
- The generic product exhibits dissolution profiles similar to those of the comparator product in buffers at pH 1.2, 4.5, and 6.8.
Class 3 Drug Products (WHO Approach)
Dosage forms of drug substances that are highly soluble and have low permeability are eligible for biowaivers under the following conditions:
- Both the reference and the generic dosage forms are very rapidly dissolving (85% dissolution in 15 minutes or less in all three media under the test conditions given above), and they do not contain any excipients and/or inactive substances that are known to alter gastrointestinal motility and/or permeability or influence drug absorption.
- Firms should show that the quantity of excipients used is consistent with the intended use. When new excipients and/or atypically large amounts of commonly used excipients are included in the dosage form, additional information documenting the absence of any significant impact on bioavailability of the drug is required.
Dissolution as a Quality Control Test and a BE Test
There is a clear difference between dissolution as a quality control test and dissolution as an in vitro equivalence (BE) test. For immediate-release dosage forms, the quality control test involves a single-point dissolution test in only one medium (generally a compendial test). On the other hand, the in vitro equivalence test (BE test) involves dissolution profile comparison in pH 1.2, 4.5, and 6.8 between the T product and the R product.