BIOAVAILABILITY, BIOEQUIVALENCE, AND DISSOLUTION
Bioavailability (BA) studies focus on determining the process and time frame by which a drug is released from the oral dosage form and moves to the site of action [see FDA Guidance Guidance for Industry—Bioavailability and Bioequivalence Studies for Orally Administered Drug Products—General Considerations (2003)]. BA is an indirect or surrogate measure of the rate and extent to which the API or active moiety is absorbed from a drug product and becomes available at its target sites of action. BA data provide an estimate of systemic drug exposure, including fraction of drug absorbed. For drug products that are not intended to be absorbed into the bloodstream, availability may be assessed by measurements that reflect the rate and extent to which the active ingredient or active moiety becomes available at the sites of action. Drug products are considered BE if a test drug product does not show a significant difference in rate and extent of absorption by comparison with a designated reference drug when administered at the same molar dose of the same active moiety in the same dosage form under similar experimental conditions in either a single dose or in multiple doses.
BA and BE generally can be obtained by serially measuring drug and/or metabolite concentrations in the systemic circulation over a prescribed period. BE studies can use other approaches when systemic drug concentrations cannot be measured or are not appropriate. For these cases, more indirect approaches to BE determination include acute pharmacodynamic endpoints, clinical endpoints, and in vitro studies that typically involve comparisons of the dissolution profiles of test and reference drug products.
BA and BE information are important in regulatory submissions. BA information broadly addresses the absorption, distribution, metabolism, and excretion of the API. For an innovator product, BE studies establish the performance of the product intended for marketing by comparing the bioavailability of the product as developed for marketing approval to the clinical trial material, the drug product used in safety/efficacy trials. For the development and regulatory approval of a generic drug product, the test drug product must be BE to the reference listed drug (RLD) product (usually the brand or innovator drug product that is designated by the applicable regulatory authority).
The ICH document titled Guidance on Q6A Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances (2000) (http://www.fda.gov/; search by document title) provides approaches for setting acceptance criteria for drug product performance. This approach relies on dissolution or disintegration based on clinically acceptable batches, as does FDA's. BE studies focus on the performance of the drug product and usually involve comparisons of two drug products: the test (T) and reference (R) or comparator product. The required studies and the determination of BE are the province of regulatory agencies. In the United States, R is termed the reference listed drug (RLD) and is so noted in FDA's Approved Drug Products with Therapeutic Equivalence Ratings [Orange Book (2008) (http://www.fda.gov/cder/ob)]. To assist countries and regions where the R product may not always be readily identifiable, WHO has prepared a document titled Annex 11 Guidance on the Selection of Comparator Pharmaceutical Products for Equivalence Assessment of Interchangeable Multisource (Generic) Products (2005) (http://www.who.int/en/; search by document title). In the WHO document, R is termed the comparator pharmaceutical product (CPP). When a country or region has a clearly defined set of CPPs, the task becomes one of requiring that a manufacturer demonstrate, to the satisfaction of its regulatory authority, that its multisource product is pharmaceutically equivalent and BE to the corresponding CPP.
BIOEQUIVALENCE
An interchangeable multisource (generic) product must be pharmaceutically equivalent (PE). The WHO document allows pharmaceutical alternatives to be considered therapeutically equivalent and interchangeable if they are BE. Further, generic products must be shown to be BE in order to be considered therapeutically equivalent (TE) to the R product (CPP). For the product to be considered PE, it must have the same active ingredient, same strength, same dosage form, same route of administration, and same labeling as the comparator product. Several methods exist to assess and document BE. These include the following:
-
Comparative pharmacokinetic studies in humans. In these studies, the active drug and/or its metabolite(s) are measured as a function of time in accessible biological fluid such as blood, plasma, serum, or urine to obtain pharmacokinetic measures such as area under the plasma drug concentration vs. time curve (AUC) and maximum concentration (Cmax) that are reflective of systemic exposure.
BE studies are designed to compare the in vivo performance of a generic product with an R product. Generally the design is a two-period, two-sequence, single-dose, crossover randomized one carried out in 18 to 36 subjects. The number of subjects should be statistically justified and not less than 12. During the study, blood samples are collected at sufficient intervals for assessing Cmax, AUC, and other parameters. Blood samples are analyzed using appropriately validated bioanalytical methodology with standard pharmacokinetic measures and statistical approaches. The statistical method for testing pharmacokinetic BE is based on the determination of the 90% confidence interval around the geometric mean ratio of the log-transformed population means (generic/R) for AUC and Cmax by carrying out two one-sided tests at the 5% level of significance.
- Other options. In addition, comparative pharmacodynamic studies in humans and comparative clinical trials can be used to document or supplement BE assessment. Beyond these clinical studies, in vitro dissolution based on the BCS can ensure BE between T and R products. In vivo documentation of equivalence is especially important for the following: narrow therapeutic range drugs; documented evidence of BE problems; modified-release pharmaceutical products designed to act by systemic absorption; and fixed-dose combination products with systemic action when at least one of the APIs requires an in vivo study.
Immediate-Release Drug Products
Single-dose, crossover BE studies are carried out at the highest dose comparing T and R products under fasting conditions. A parallel study design can be used for drugs that have a very long elimination half-life (t½). Sampling truncation at 72 hours may be allowable by regulatory agencies. Lower strength(s) of the dosage form can be given a biowaiver based on dosage form proportionality and dissolution profile similarity. Food-effect studies are required if there is an indication in the labeling that concomitant administration of food may diminish, increase, or not influence the BA of the drug product.
Modified-Release Drug Products
BE studies for extended-release dosage forms are carried out as single-dose, crossover studies under fasting and fed conditions at the highest dose to compare T and R products. A single-dose study is more sensitive than multiple-dose, steady-state studies in assessing in vivo drug product performance, particularly with regard to the phenomenon of dose dumping, i.e., the rapid and unintended premature release of the active ingredient from an extended-release product into the bloodstream. Lower strengths of an extended-release dosage form may not require an in vivo study based on use of the same drug-releasing mechanism, dosage form proportionality, and similar dissolution profile.
Orally Administered Drug Products, Not for Systemic Effect
Some oral drug products are intended for local activity. Mesalamine and cholestyramine are examples of drugs that are intended for local activity. For these types of drugs, systemic absorption from the gastrointestinal tract is minimal; thus a comparative clinical trial is required while a systemic drug exposure profile also may be required. In some cases, in vitro studies may be appropriate; such as including comparison of cholestyramine binding to bile salts.
Bioequivalence Studies
Objective—
The objective of a BE study is to measure and compare formulation performance between two or more pharmaceutically equivalent drug products. Drug availability from T and R products should not be statistically different when the drug is administered to patients or subjects at the same molar dose under similar experimental conditions.
Design—
The design of a BE study depends on the objectives of the study, the ability to analyze the drug (and metabolites) in biological fluids, the pharmacodynamics of the drug substance, the route of drug administration, and the nature of the drug and drug product. Pharmacokinetic parameters, pharmacodynamic parameters, clinical observations, and/or in vitro studies may be used to determine drug BA from a drug product.
Some possible BE study designs include the following:
- Single-dose, two-way crossover study under fasted conditions
- Single-dose, two-way crossover study under fed conditions
- Single-dose, parallel study under fasted conditions
- Single-dose, replicate design
- Single-dose, partial replicate design
- Multiple-dose, two-way crossover study, fasted conditions
- Pharmacodynamic or clinical endpoint study
- In vitro dissolution profile comparisons
The standard BE study is a crossover design (e.g., Latin square crossover design) in which each subject receives the test drug product and the reference product on separate occasions. Studies are usually evaluated by a single-dose, two-period, two-treatment, two-sequence, open-label, randomized crossover design comparing equal doses of the test and reference products in fasted or fed adult healthy subjects. A multiple-dose study may be required for some extended-release drug products. A washout period is scheduled between the two periods to allow the subjects to completely eliminate the drug absorbed from the first dose before administration of the second dose. If the predose concentration is 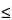 5% of the Cmax value in that subject, the subject's data without any adjustments can be included in all pharmacokinetic measurements and calculations. Samples of an accessible biologic fluid such as blood characterize the drug concentration vs. time profile. During the fasting study subjects are fasted at least 10 hours. A pre-dose (0 time) blood sample is taken. The drug product is given with 240 mL (8 fluid ounces) of water. No food is allowed for at least 4 hours post-dose. Blood sampling is performed periodically after dose administration according to protocol. A food intervention or food effect study is conducted with standard meal conditions that are expected to provide the greatest effects on gastrointestinal physiology so that systemic drug availability is maximally affected. In addition, the high lipid content of the meal may affect the rate of drug release from the product, in situ. A high-fat (approximately 50% of total caloric content of the meal) and high-calorie (approximately 800 to 1000 calories) meal is recommended as a test meal for food-effect BA and fed BE studies. This test meal should derive approximately 150, 250, and 500–600 calories from protein, carbohydrate, and fat, respectively. The drug product is given with 240 mL (8 fluid ounces) of water after ingestion of the standard meal. Subjects should consume identical meals at the same time during a testing period.
5% of the Cmax value in that subject, the subject's data without any adjustments can be included in all pharmacokinetic measurements and calculations. Samples of an accessible biologic fluid such as blood characterize the drug concentration vs. time profile. During the fasting study subjects are fasted at least 10 hours. A pre-dose (0 time) blood sample is taken. The drug product is given with 240 mL (8 fluid ounces) of water. No food is allowed for at least 4 hours post-dose. Blood sampling is performed periodically after dose administration according to protocol. A food intervention or food effect study is conducted with standard meal conditions that are expected to provide the greatest effects on gastrointestinal physiology so that systemic drug availability is maximally affected. In addition, the high lipid content of the meal may affect the rate of drug release from the product, in situ. A high-fat (approximately 50% of total caloric content of the meal) and high-calorie (approximately 800 to 1000 calories) meal is recommended as a test meal for food-effect BA and fed BE studies. This test meal should derive approximately 150, 250, and 500–600 calories from protein, carbohydrate, and fat, respectively. The drug product is given with 240 mL (8 fluid ounces) of water after ingestion of the standard meal. Subjects should consume identical meals at the same time during a testing period.
Analysis of Samples—
Samples, usually plasma, are analyzed for the active drug and, on occasion, active metabolite concentrations by a validated bioanalytical method.
Pharmacokinetic Parameters—
Pharmacokinetic parameters are obtained from the resulting concentration-time curves. Two major pharmacokinetic parameters are used to assess the rate and extent of systemic drug absorption. AUC reflects the extent of drug absorption, and the peak drug concentration (Cmax) reflects the rate of drug absorption. Other pharmacokinetic parameters may include the time to peak drug concentration (Tmax), the elimination rate constant (k), elimination half-life (t½), lag time (Tlag), and others.
Statistical Analysis
Pharmacokinetic parameters are analyzed statistically to determine whether the T and R products yield comparable values. Because BE studies may use small sample sizes, log transformation of the data allows the frequency distribution of the data to be more normalized so that parametric statistical analyses may be performed (FDA, Guidance for Industry: Statistical Approaches to Establishing Bioequivalence (2001) (http://www.fda.gov/; search by document title).
Parametric (normal-theory) general linear model procedures are recommended for the analysis of pharmacokinetic data derived from in vivo BE studies. An analysis of variance (ANOVA) should be performed on the pharmacokinetic parameters AUC and Cmax using appropriate statistical programs and models. For example, for a conventional two-treatment, two-period, two-sequence (2 × 2) randomized crossover study design, the statistical model often includes factors accounting for the following sources of variation:
- Sequence (sometimes called Group or Order)
- Subjects, nested in sequences
- Period (or Phase)
- Treatment (sometimes called Drug or Formulation)
The sequence effect should be tested using the [subject (sequence)] mean square from the ANOVA as an error term. All other main effects should be tested against the residual error (error mean square) from the ANOVA. The least-squares means (LSMEANS) statement should be used to calculate least-squares means for treatments. Estimates should be obtained for the adjusted differences between treatment means and the standard error associated with these differences.
The statistical assumptions underlying the ANOVA are as follows:
- Randomization of samples
- Homogeneity of variances
- Additivity (linearity) of the statistical model
- Independence and normality of residuals
In BE studies, these assumptions can be interpreted as follows:
- The subjects chosen for the study should be randomly assigned to the sequences of the study.
- The variances associated with the two treatments, as well as between the sequence groups, should be equal or at least comparable.
- The main effects of the statistical model, such as subject, sequence, period, and treatment effect for a standard 2 × 2 crossover study, should be additive. There should be no interactions between these effects.
- The residuals of the model should be independently and normally distributed.
If these assumptions are not met, additional steps should be taken prior to the ANOVA, including data transformation to improve the fit of the assumptions or use of a nonparametric statistical test in place of ANOVA. However, the normality and constant variance assumptions in the ANOVA model are known to be relatively robust (i.e., a small or moderate departure from each, or both of these assumptions, will not have a significant effect on the final result). The rationale for log transformation is provided in FDA's Guidance Statistical Approaches to Establishing Bioequivalence. Justification should be provided if untransformed data is to be used.
The Two One-Sided Tests Procedure—
A testing procedure termed the two one-sided tests procedure is used to determine the comparability of geometric mean values for pharmacokinetic parameters measured after administration of the test and reference products.1 The two one-sided tests procedure decides whether T is not importantly less than R and whether R is not importantly less than T. Most often, 20% defines an important difference. The statistical procedure involves the calculation of a confidence interval for the ratio (or difference) between T and R pharmacokinetic variable averages. The limits of the observed confidence interval must fall within a predetermined range for the ratio (or difference) of the product averages. Point estimate mean ratios (T/R) derived from the log-transformed AUC and Cmax data must be between 80% and 125%. Because data are log transformed, T/R = 80/100 = 80% and R/T = 100/80 = 125%. In addition, the 90% confidence intervals for the geometric mean ratios (T/R) for AUC and Cmax must be between 80% and 125%. The regulatory requirements for the range of 90% confidence intervals for Cmax may be different in countries outside the United States.
Bio-Inequivalence—
The failure to demonstrate BE may be due to a performance failure of the T product or to an inadequate study design. The failure to demonstrate BE due to an inadequate study design can be due to improper sampling in which (1) the sampling time for Cmax was not properly obtained or (2) the number of samples taken did not adequately describe the plasma drug concentration vs. time profile. Often with highly variable drugs (e.g., %CV >30%), too few subjects were used in the study, and therefore the study was not powered adequately.
Presentation of Data.
The drug concentration in biological fluid at each sampling time point should be furnished untransformed for all the subjects who participated in the study. The derived pharmacokinetic parameters also should be furnished untransformed. The mean, the standard deviation, and the coefficient of variation (CV) for each variable should be computed and tabulated in the final report.
To facilitate BE comparisons, pharmacokinetic parameters for each individual should be displayed in parallel for the formulations tested. In particular, for AUC and Cmax, the difference (T – R), the ratio (T/R), and the log of ratio (log T/R or ln T/R) between the T and R values should be tabulated side by side for all the subjects. For each subject, the summary tables should indicate in which sequence (T, R or R, T) the subject received the product. Histograms showing the frequency distribution of the difference and ln ratio (or log ratio) for the major pharmacokinetic parameters (AUC and Cmax) are useful in the submission.
In addition to the arithmetic mean for the T and R products, the geometric means (antilog of the means of the logs), means of the logs, and standard deviations of the logs should be calculated for AUC and Cmax. All means, including arithmetic mean, geometric mean, and means of the logs, as well as standard deviations and CVs, should be included in the report.