Heparin Sodium
Change to read:
» Heparin Sodium is the sodium salt of sulfated glycosaminoglycans present as a mixture of heterogeneous molecules varying in molecular weights. It is present in mammalian tissues and is usually obtained from the intestinal mucosa or other suitable tissues of domestic mammals used for food by man.  The sourcing of heparin material must be specified in compliance with applicable regulatory requirements. The manufacturing process must be validated to demonstrate clearance and inactivation of relevant infectious and adventitious agents (e.g., viruses, TSE agents). See Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin
The sourcing of heparin material must be specified in compliance with applicable regulatory requirements. The manufacturing process must be validated to demonstrate clearance and inactivation of relevant infectious and adventitious agents (e.g., viruses, TSE agents). See Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin  1050
1050 for general guidance on viral safety evaluation. (RB 18-Jun-2008) It is purified to retain a combination of activities against different fractions of the blood clotting sequence. It is composed of polymers of alternating derivatives of
for general guidance on viral safety evaluation. (RB 18-Jun-2008) It is purified to retain a combination of activities against different fractions of the blood clotting sequence. It is composed of polymers of alternating derivatives of 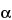 - (RB 18-Jun-2008) d-glucosamine (N-sulfated, O-sulfated, or N-acetylated) and uronic acid (- (RB 18-Jun-2008) l-iduronic acid or
- (RB 18-Jun-2008) d-glucosamine (N-sulfated, O-sulfated, or N-acetylated) and uronic acid (- (RB 18-Jun-2008) l-iduronic acid or 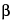 - (RB 18-Jun-2008) d-glucuronic acid) joined by glycosidic linkages. The component activities of the mixture are in ratios corresponding to those shown by the USP Heparin Sodium Reference Standard. Some of these components have the property of prolonging the clotting time of blood. This occurs mainly through the formation of a complex of each component with the plasma proteins antithrombin III and heparin cofactor II to potentiate the inactivation of thrombin. Other coagulation proteases in the clotting sequence, such as activated factor X, are also inhibited. The potency of Heparin Sodium, calculated on the dried basis, is not less than 140 USP Heparin Units in each mg, and not less than 90.0 percent and not more than 110.0 percent of the potency stated on the label.
- (RB 18-Jun-2008) d-glucuronic acid) joined by glycosidic linkages. The component activities of the mixture are in ratios corresponding to those shown by the USP Heparin Sodium Reference Standard. Some of these components have the property of prolonging the clotting time of blood. This occurs mainly through the formation of a complex of each component with the plasma proteins antithrombin III and heparin cofactor II to potentiate the inactivation of thrombin. Other coagulation proteases in the clotting sequence, such as activated factor X, are also inhibited. The potency of Heparin Sodium, calculated on the dried basis, is not less than 140 USP Heparin Units in each mg, and not less than 90.0 percent and not more than 110.0 percent of the potency stated on the label.
note—The USP Heparin Unit is defined by the USP Heparin Sodium Reference Standard and can be independent of International Units. The respective units are not equivalent (see General Notices). The Unit for Anti-Factor Xa activity is defined by the USP Heparin Sodium Reference Standard and is equivalent in potency to that Standard.
Packaging and storage—
Preserve in tight containers, and store below 40 , preferably at room temperature.
, preferably at room temperature.
Labeling—
Label it to indicate the tissue and the animal species from which it is derived.
Change to read:
USP Reference standards USP Endotoxin RS.
USP Heparin Sodium RS.
USP Heparin Sodium System Suitability RS
 .
.
USP Heparin Sodium Identification RS.
Change to read:
Identification—
B:
1H NMR spectrum (see Nuclear Magnetic Resonance 761)—
Standard solution—
Prepare a solution of USP Heparin Sodium Identification RS at not less than 12 mg per mL in deuterium oxide.
System suitability solution—
Prepare a solution of USP Heparin Sodium System Suitability RS at not less than 12 mg per mL in deuterium oxide.
Test solution—
Prepare a solution containing not less than 12 mg of Heparin Sodium per mL in deuterium oxide.
Procedure—
Using a pulsed (Fourier transform) NMR spectrometer operating at not less than 500 MHz for 1H, acquire a free induction decay (FID). Record the 1H NMR spectra of the Standard solution and the System suitability solution at 25: the number of transients is adjusted until the signal-to-noise ratio of the N-acetyl heparin signal in the Standard solution is at least 200/1 in the region near 2 ppm. The Standard solution must be run at least daily when Test solutions are being run. For the Standard solution, the chemical shift corresponding to N-acetyl protons of heparin must be set at 2.04 ppm. The chemical shifts for heparin and over-sulfated chondroitin sulfate in the system suitability standard is observed at 2.04 ± 0.02 and 2.16 ± 0.03 ppm, respectively. Record the 1H NMR spectrum of the Test solution at 25. The N-acetyl protons of heparin must show a major signal at 2.04 ± 0.02 ppm. A signal, corresponding to N-acetyl protons of dermatan sulfate, may show near 2.08 ppm. Observation of a resonance at 2.16 ± 0.03 ppm would indicate the presence of over-sulfated chondroitin sulfate. Over-sulfated chondroitin sulfate has no other resonances between 2.12 and 3.00 ppm.
C:
For heparin sodium of porcine origin (see Biotechnology-Derived Articles—Capillary Electrophoresis 1053)—
Standard solution—
Prepare a solution of USP Heparin Sodium Identification RS in water having a concentration of 2 mg per mL.
System suitability solution—
Prepare a solution of USP Heparin Sodium System Suitability RS in water having a concentration of 2 mg per mL.
Test solution—
Reconstitute an accurately weighed quantity of Heparin Sodium in water to obtain a solution that is 2 mg per mL. Filter the solution if necessary.
Capillary electrophoresis buffer—
Transfer 1.0 g of monobasic sodium phosphate monohydrate to a beaker, and add 195 mL of water. Adjust with phosphoric acid to a pH of 3.5. Transfer the solution into a 200-mL volumetric flask, and dilute with water to volume. Filter the buffer with a 0.2-µm membrane filter. Degas the buffer before use if necessary.
Electrophoretic system—
[Note—Based on instrument requirements, the field applied across the capillary and the conditions for the sample injection may be varied to achieve system suitability.] The capillary electropherograph is equipped with a 200-nm detector and a 60- to 65-cm (50- to 56-cm effective length) uncoated fused silica capillary, with an internal diameter of 50 µm, with the temperature controlled at 25. Apply a field strength of 465.1 V per cm for 15 minutes, using the Capillary electrophoresis buffer as the electrolyte in both buffer reservoirs. Electropherograph the USP Heparin Sodium System Suitability RS, using a 30-sec injection at 0.7 psi, and record the peak responses as directed for Procedure: the migration time for heparin sodium and over-sulfated chondroitin sulfate differs by not less than 4.0% of the heparin migration time, with over-sulfated chondroitin sulfate always migrating faster than heparin sodium. The baseline is stable. Rinse the capillary with 0.1 M phosphoric acid for at least 0.5 minutes at 40 psi, followed by water for at least 2 minutes at 40 psi, and then by the Capillary electrophoresis buffer for at least 2 minutes at 40 psi between injections.
Procedure—
Electropherograph the Standard solution and the Test solution using a 30-sec injection at 0.7 psi into the anodic end of the capillary, and record the electropherograms. The electropherogram of the System suitability solution is comparable to a typical electropherogram provided with the USP Heparin Sodium System Suitability RS. The electropherogram of the Test solution is similar to that of the Standard solution, and does not exhibit a sharp distinguishable peak that is less than one minute in front of the main heparin peak.
D: (RB 18-Jun-2008)
It meets the requirements of the flame test for Sodium 191.
Bacterial endotoxins 85—
It contains not more than 0.03 USP Endotoxin Unit per USP Heparin Unit.
Sterility 71 (where it is labeled as sterile):
meets the requirements.
pH 791:
between 5.0 and 7.5, in a solution (1 in 100).
Loss on drying 731—
Dry it in vacuum at 60 for 3 hours: it loses not more than 5.0% of its weight.
Residue on ignition 281:
between 28.0% and 41.0%.
Protein—
To 1 mL of a solution (1 in 100) add 5 drops of trichloroacetic acid solution (1 in 5): no precipitate or turbidity forms.
Heavy metals, Method II 231:
0.003%.
Anti-factor Xa activity—
pH 8.4 Buffer—
Dissolve amounts of tris(hydroxymethyl)aminomethane, edetic acid, and sodium chloride in water containing 0.1% of polyethylene glycol 6000 to obtain a solution having concentrations of 0.050 M, 0.0075 M, and 0.175 M, respectively. Adjust, if necessary, with hydrochloric acid or sodium hydroxide solution to a pH of 8.4.
Antithrombin III solution—
Reconstitute an accurately weighed quantity of antithrombin III (see Reagent Specifications under Reagents, Indicators, and Solutions) in pH 8.4 Buffer to obtain a solution having a concentration of 1.0 Antithrombin III Unit per mL.
Factor Xa solution—
Reconstitute an accurately weighed quantity of bovine factor Xa (see Factor Xa in Reagent Specifications under Reagents, Indicators, and Solutions) in pH 8.4 Buffer to obtain a solution that gives an absorbance value between 0.65 and 1.25 at 405 nm when assayed as described below but using 30 µL of pH 8.4 Buffer instead of 30 µL of the Standard solutions or the Test solutions.
note—Factor Xa solution contains about 3 nanokatalytic units per mL, but can vary depending upon the manufacturer of factor Xa or the substrate used.
Chromogenic substrate solution—
Prepare a solution of a suitable chromogenic substrate for amidolytic test (see Reagent Specifications under Reagents, Indicators, and Solutions) specific for factor Xa in water to obtain a concentration of about 1 mM.
Stopping solution—
Prepare a 20% (v/v) solution of acetic acid in water.
Standard solutions—
Dilute an accurately measured volume of USP Heparin Sodium RS with pH 8.4 Buffer to obtain at least 5 (out of 7 below) solutions having known activities of about 0.375, 0.3125, 0.25, 0.188, 0.125, 0.0625, and 0.0313 USP Heparin Unit per mL.
Test solutions—
Dissolve or dilute an accurately measured quantity of Heparin Sodium in pH 8.4 Buffer, and dilute with the same buffer to obtain solutions having activities approximately equal to those of the Standard solutions.
Procedure—
[note—Perform the test with each Standard solution and Test solution in duplicate.] To each of a series of suitable plastic tubes placed in a water bath set at 37, transfer 120 µL of pH 8.4 Buffer. Then separately transfer 30 µL of the different dilutions of the Standard solutions or the Test solutions to the tubes. Add 150 µL of Antithrombin III solution, prewarmed at 37 for 15 minutes, to each tube, mix, and incubate for 2 minutes. Add 300 µL of Factor Xa solution, prewarmed at 37 for 15 minutes, to each tube, mix, and incubate for 2 minutes. Add 300 µL of Chromogenic substrate solution, prewarmed at 37 for 15 minutes, to each tube, mix, and incubate for exactly 2 minutes. Add 150 µL of Stopping solution to each tube, and mix. Prepare a blank for zeroing the spectrophotometer by adding the reagents in reverse order, starting with the Stopping solution and ending with the addition of 150 µL of pH 8.4 Buffer, and excluding the Standard solutions or the Test solutions. Record the absorbance at 405 nm against the blank.
Calculations—
Plot the log of the absorbance values of the Standard solutions and the Test solutions versus heparin concentrations in USP Units. Construct separate straight lines of best fit using least-squares linear regression analyses for the Standard solutions and the Test solutions, and determine the slope for each regression line. Calculate the potency of Heparin Sodium by the formula:
P(ST / SS)
in which P is the potency of USP Heparin Sodium RS; and ST and SS are the slopes of the lines from the Test solutions and the Standard solutions, respectively. Express the Anti-factor Xa potency of the Test solution as a percentage of the heparin concentration determined in the Assay. Calculate the percentage of anti-factor Xa activity against anticoagulant activity by the formula:
100(anti-factor Xa potency / anticoagulant potency)
Not less than 80% and not more than 120% is found.
Nitrogen content, Method I 461:
between 1.3% and 2.5%, calculated on the dried basis, the procedure for Nitrates and Nitrates Absent being used.
Assay—
Standard preparation—
Determine by preliminary trial, if necessary, approximately the minimum quantity of USP Heparin Sodium RS which, when added in 0.8 mL of saline TS, maintains fluidity in 1 mL of prepared plasma for 1 hour after the addition of 0.2 mL of calcium chloride solution (1 in 100). This quantity is usually between 1 and 3 USP Heparin Units. On the day of the assay prepare a Standard preparation such that it contains, in each 0.8 mL of saline TS, the above-determined quantity of the Reference Standard.
Assay preparation—
Dissolve about 25 mg of Heparin Sodium, accurately weighed, in sufficient saline TS to give a concentration of 1 mg per mL, and dilute quantitatively to a concentration estimated to correspond to that of the Standard preparation.
Preparation of plasma—
Collect blood from sheep directly into a vessel containing 8% sodium citrate solution in the proportion of one volume to each 19 volumes of blood to be collected. Mix immediately by gentle agitation and inversion of the vessel. Promptly centrifuge the blood, and pool the separated plasma. To a 1-mL portion of the pooled plasma in a clean test tube add 0.2 mL of calcium chloride solution (1 in 100), and mix. Consider the plasma suitable for use if a solid clot forms within 5 minutes. To store plasma for future use, subdivide the pooled lot into portions not exceeding 100 mL in volume, and store in the frozen state, preventing even partial thawing prior to use. For use in the assay, thaw the frozen plasma in a water bath at a temperature not exceeding 37. Remove particulate matter by straining the thawed plasma through a coarse filter.
Procedure—
To meticulously clean 13-mm × 100-mm test tubes add graded amounts of the Standard preparation, selecting the amounts so that the largest does not exceed 0.8 mL and so that they correspond roughly to a geometric series in which each step is approximately 5% greater than the next lower. To each tube so prepared add sufficient saline TS to make the total volume 0.8 mL. Add 1.0 mL of prepared plasma to each tube. Then add 0.2 mL of calcium chloride solution (1 in 100), note the time, immediately insert a suitable stopper in each tube, and mix the contents by inverting three times in such a way that the entire inner surface of the tube is wet.
In the same manner set up a series using the Assay preparation, completing the entire process of preparing and mixing the tubes of both the Standard preparation and the Assay preparation within 20 minutes after the addition of the prepared plasma. One hour, accurately timed, after the addition of the calcium chloride, determine the extent of clotting in each tube, recognizing three grades (0.25, 0.50, and 0.75) between zero and full clotting (1.0). If the series does not contain 2 tubes graded more than 0.5 and 2 tubes graded less than 0.5, repeat the assay, using appropriately modified Standard preparation and Assay preparation.
Calculation—
Convert to logarithms the volumes of Standard preparation used in the successive 5 or 6 tubes that bracket a grade of clotting of 0.5, including at least 2 tubes with a larger and 2 tubes with a smaller grade than 0.5. Number and list the tubes serially, and tabulate for each the grade of clotting observed in each tube. From the log-volumes, x, and separately from their corresponding grades of clotting, y, compute the paired averages xi and yi of Tubes 1, 2, and 3, of Tubes 2, 3, and 4, of Tubes 3, 4, and 5, and, where the series consists of 6 tubes, of Tubes 4, 5, and 6, respectively. If for one of these paired averages the average grade, yi, is exactly 0.50, the corresponding xi is the median log-volume of the Standard preparation, xS. Otherwise, interpolate xS from the paired values of yi, xi and yi+1, xi+1 that fall immediately below and above grade 0.5 as
xS = xi + (yi 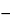 0.5)(xi+1 xi) / (yi yi+1)
0.5)(xi+1 xi) / (yi yi+1)
From the paired data on the tubes of the Assay preparation, compute similarly its median log-volume xU.
The log potency of the Assay preparation is
M = xS xU + log R
where R = vS /vU is the ratio of the USP Heparin Units (vS) per mL of the Standard preparation to the mg (vU) of Heparin Sodium per mL of the Assay preparation.
Repeat the assay independently, and average the two or more values of M to obtain M. If the second determination of M differs by more than 0.05 from the first determination, continue the assay until the log confidence interval computed as directed under Confidence Intervals for Individual Assays in Design and Analysis of Biological Assays 111 does not exceed 0.20. The potency of Heparin Sodium in USP Heparin Units per mg is P* = antilog M.
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
| Monograph | Anita Y. Szajek, Ph.D.
Senior Scientist 1-301-816-8325 |
(BBBBP05) Biologics and Biotechnology - Blood and Blood Products |
| Reference Standards | Lili Wang, Technical Services Scientist 1-301-816-8129 RSTech@usp.org |
|
| Radhakrishna S Tirumalai, Ph.D.
Senior Scientist 1-301-816-8339 |
(MSA05) Microbiology and Sterility Assurance | |
| Radhakrishna S Tirumalai, Ph.D.
Senior Scientist 1-301-816-8339 |
(MSA05) Microbiology and Sterility Assurance |
USP32–NF27 Page 2552
Pharmacopeial Forum: Volume No. 33(2) Page 238