note—Inasmuch as this chapter is for purposes of general information only, no statement in the chapter is intended to modify or supplant any of the specific requirements pertinent to Pharmacopeial articles, which are given elsewhere in this Pharmacopeia.
Aspects of drug product stability that are of primary concern to the pharmacist in the dispensing of medications are discussed herein.
Pharmacists should avoid ingredients and conditions that could result in excessive physical deterioration or chemical decomposition of drug preparations, especially when compounding (see Pharmaceutical Compounding—Nonsterile Preparations  795
795 ). The stability and clinical effect of manufactured dosage forms can be greatly compromised by seemingly negligible alterations or inappropriate prescription compounding. Pharmacists should establish and maintain compounding conditions that include the ensuring of drug stability to help prevent therapeutic failure and adverse responses.
). The stability and clinical effect of manufactured dosage forms can be greatly compromised by seemingly negligible alterations or inappropriate prescription compounding. Pharmacists should establish and maintain compounding conditions that include the ensuring of drug stability to help prevent therapeutic failure and adverse responses.
Stability—Stability is defined as the extent to which a product retains, within specified limits, and throughout its period of storage and use (i.e., its shelf-life), the same properties and characteristics that it possessed at the time of its manufacture. Five types of stability generally recognized are shown in the accompanying table.
Criteria for Acceptable Levels of Stability
| Type of Stability |
Conditions Maintained Throughout the Shelf Life of the Drug Product |
| Chemical | Each active ingredient retains its chemical integrity and labeled potency, within the specified limits. |
| Physical | The original physical properties, including appearance, palatability, uniformity, dissolution, and suspendability, are retained. |
| Microbiological | Sterility or resistance to microbial growth is retained according to the specified requirements. Antimicrobial agents that are present retain effectiveness within the specified limits. |
| Therapeutic | The therapeutic effect remains unchanged. |
| Toxicological | No significant increase in toxicity occurs. |
FACTORS AFFECTING PRODUCT STABILITY
Each ingredient, whether therapeutically active or pharmaceutically necessary, can affect the stability of drug substances and dosage forms. The primary environmental factors that can reduce stability include exposure to adverse temperatures, light, humidity, oxygen, and carbon dioxide. The major dosage form factors that influence drug stability include particle size (especially in emulsions and suspensions), pH, solvent system composition (i.e., percentage of “free” water and overall polarity), compatibility of anions and cations, solution ionic strength, primary container, specific chemical additives, and molecular binding and diffusion of drugs and excipients. In dosage forms, the following reactions usually cause loss of active drug content, and they usually do not provide obvious visual or olfactory evidence of their occurrence.
Hydrolysis—
Esters and 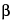 -lactams are the chemical bonds that are most likely to hydrolyze in the presence of water. For example, the acetyl ester in aspirin is hydrolyzed to acetic acid and salicylic acid in the presence of moisture, but in a dry environment the hydrolysis of aspirin is negligible. The aspirin hydrolysis rate increases in direct proportion to the water vapor pressure in an environment.
-lactams are the chemical bonds that are most likely to hydrolyze in the presence of water. For example, the acetyl ester in aspirin is hydrolyzed to acetic acid and salicylic acid in the presence of moisture, but in a dry environment the hydrolysis of aspirin is negligible. The aspirin hydrolysis rate increases in direct proportion to the water vapor pressure in an environment.
The amide bond also hydrolyzes, though generally at a slower rate than comparable esters. For example, procaine (an ester) will hydrolyze upon autoclaving, but procainamide will not. The amide or peptide bond in peptides and proteins varies in the lability to hydrolysis.
The lactam and azomethine (or imine) bonds in benzodiazepines are also labile to hydrolysis. The major chemical accelerators or catalysts of hydrolysis are adverse pH and specific chemicals (e.g., dextrose and copper in the case of ampicillin hydrolysis).
Epimerization—
Members of the tetracycline family are most likely to incur epimerization. This reaction occurs rapidly when the dissolved drug is exposed to a pH of an intermediate range (higher than 3), and it results in the steric rearrangement of the dimethylamino group. The epimer of tetracycline, epitetracycline, has little or no antibacterial activity.
Decarboxylation—
Some dissolved carboxylic acids, such as p-aminosalicylic acid, lose carbon dioxide from the carboxyl group when heated. The resulting product has reduced pharmacological potency.
-Keto decarboxylation can occur in some solid antibiotics that have a carbonyl group on the -carbon of a carboxylic acid or a carboxylate anion. Such decarboxylations will occur in the following antibiotics: carbenicillin sodium, carbenicillin free acid, ticarcillin sodium, and ticarcillin free acid.
Dehydration—
Acid-catalyzed dehydration of tetracycline forms epianhydrotetracycline, a product that both lacks antibacterial activity and causes toxicity.
Oxidation—
The molecular structures most likely to oxidize are those with a hydroxyl group directly bonded to an aromatic ring (e.g., phenol derivatives such as catecholamines and morphine), conjugated dienes (e.g., vitamin A and unsaturated free fatty acids), heterocyclic aromatic rings, nitroso and nitrite derivatives, and aldehydes (e.g., flavorings). Products of oxidation usually lack therapeutic activity. Visual identification of oxidation, for example, the change from colorless epinephrine to its amber colored products, may not be visible in some dilutions or to some eyes.
Oxidation is catalyzed by pH values that are higher than optimum, polyvalent heavy metal ions (e.g., copper and iron), and exposure to oxygen and UV illumination. The latter two causes of oxidation justify the use of antioxidant chemicals, nitrogen atmospheres during ampul and vial filling, opaque external packaging, and transparent amber glass or plastic containers.
Photochemical Decomposition—
Exposure to, primarily, UV illumination may cause oxidation (photo-oxidation) and scission (photolysis) of covalent bonds. Nifedipine, nitroprusside, riboflavin, and phenothiazines are very labile to photo-oxidation. In susceptible compounds, photochemical energy creates free radical intermediates, which can perpetuate chain reactions.
Ionic Strength—
The effect of the total concentration of dissolved electrolytes on the rate of hydrolysis reactions results from the influence of ionic strength on interionic attraction. In general, the hydrolysis rate constant is inversely proportional to the ionic strength with oppositely charged ions (e.g., drug cation and excipient anions) and directly proportional to the ionic strength with ions of like charge. A reaction that produces an ion of opposite charge to the original drug ion because of the increasing ionic strength, can increase the drug hydrolysis rate as the reaction proceeds. High ionic strength of inorganic salts can also reduce the solubility of some other drugs.
pH Effect—
The degradation of many drugs in solution accelerates or decelerates exponentially as the pH is decreased or increased over a specific range of pH values. Improper pH ranks with exposure to elevated temperature as a factor most likely to cause a clinically significant loss of drug, resulting from hydrolysis and oxidation reactions. A drug solution or suspension, for example, may be stable for days, weeks, or even years in its original formulation, but when mixed with another liquid that changes the pH, it degrades in minutes or days. It is possible that a pH change of only 1 unit (e.g., from 4 to 3 or 8 to 9) could decrease drug stability by a factor of 10 or greater.
A pH buffer system, which is usually a weak acid or base and its salt, is a common excipient used in liquid preparations to maintain the pH in a range that minimizes the drug degradation rate. The pH of drug solutions may also be either buffered or adjusted to achieve drug solubility. For example, pH in relation to pKa controls the fractions of the usually more soluble ionized and less soluble nonionized species of weak organic electrolytes.
The influence of pH on the physical stability of two phase systems, especially emulsions, is also important. For example, intravenous fat emulsion is destabilized by acidic pH.
Interionic (IonN+–IonN–) Compatibility—
The compatibility or solubility of oppositely charged ions depends mainly on the number of charges per ion and the molecular size of the ions. In general, polyvalent ions of opposite charge are more likely to be incompatible. Thus, an incompatibility is likely to occur upon the addition of a large ion with a charge opposite to that of the drug.
Solid State Stability—
Solid state reactions are relatively slow; thus, stability of drugs in the solid state is rarely a dispensing concern. The degradation rate of dry solids is usually characterized by first-order kinetics or a sigmoid curve. Therefore, solid drugs with lower melting point temperatures should not be combined with other chemicals that would form a eutectic mixture.
When moisture is present, the solid drug decomposition may change to zero-order chemical kinetics because the rate is controlled by the relatively small fraction of the drug that exists in a saturated solution, which is located (usually imperceptibly) at the surface or in the bulk of the solid drug product.
Temperature—
In general, the rate of a chemical reaction increases exponentially for each 10 increase in temperature. This relationship has been observed for nearly all drug hydrolysis and some drug oxidation reactions. The actual factor of rate increase depends on the activation energy of the particular reaction. The activation energy is a function of the specific reactive bond and the drug formulation (e.g., solvent, pH, additives). As an example, consider a hydrolyzable drug that is exposed to a 20 increase in temperature, such as that from cold to controlled room temperature (see General Notices and Requirements). The shelf life of the drug at controlled room temperature should be expected to decrease to one-fourth to one-twenty-fifth of its shelf life under refrigeration.
increase in temperature. This relationship has been observed for nearly all drug hydrolysis and some drug oxidation reactions. The actual factor of rate increase depends on the activation energy of the particular reaction. The activation energy is a function of the specific reactive bond and the drug formulation (e.g., solvent, pH, additives). As an example, consider a hydrolyzable drug that is exposed to a 20 increase in temperature, such as that from cold to controlled room temperature (see General Notices and Requirements). The shelf life of the drug at controlled room temperature should be expected to decrease to one-fourth to one-twenty-fifth of its shelf life under refrigeration.
The pharmacist should also be aware that inappropriately cold temperatures may cause harm. For example, refrigeration may cause extreme viscosity in some liquid drugs and cause supersaturation in others. Freezing may either break or cause a large increase in the droplet size of emulsions; it can denature proteins; and in rare cases, it can cause less soluble polymorphic states of some drugs to form.
STABILITY STUDIES IN MANUFACTURING
The scope and design of a stability study vary according to the product and the manufacturer concerned. Ordinarily the formulator of a product first determines the effects of temperature, light, air, pH, moisture, trace metals, and commonly used excipients or solvents on the active ingredient(s). From this information, one or more formulations of each dosage form are prepared, packaged in suitable containers, and stored under a variety of environmental conditions, both exaggerated and normal. See Pharmaceutical Stability 1150. At appropriate time intervals, samples of the product are assayed for potency by use of a stability-indicating method, observed for physical changes, and, where applicable, tested for sterility and or for resistance to microbial growth and for toxicity and bioavailability. Such a study, in combination with clinical and toxicological results, enables the manufacturer to select the optimum formulation and container and to assign recommended storage conditions and an expiration date for each dosage form in its package.
Responsibility of Pharmacists
Pharmacists help to ensure that the products under their supervision meet acceptable criteria of stability by (1) dispensing oldest stock first and observing expiration dates, (2) storing products under the environmental conditions stated in the individual monographs, labeling, or both, (3) observing products for evidence of instability, (4) properly treating and labeling products that are repackaged, diluted, or mixed with other products, (5) dispensing in the proper container with the proper closure, and (6) informing and educating patients concerning the proper storage and use of the products, including the disposition of outdated or excessively aged prescriptions.
Rotation of Stock and Observance of Expiration Dates—
Proper rotation of stock is necessary to ensure the dispensing of suitable products. A product that is dispensed infrequently should be closely monitored so that old stocks are given special attention, particularly with regard to expiration dates. The manufacturer can guarantee the quality of a product up to the time designated as its expiration date only if the product has been stored in the original container under recommended storage conditions.
Storage under Recommended Environmental Conditions—
In most instances, the recommended storage conditions are stated on the label, in which case it is imperative to adhere to those conditions. They may include a specified temperature range or a designated storage place or condition (e.g., “refrigerator,” or “controlled room temperature”) as defined in the General Notices. Supplemental instructions, such as a direction to protect the product from light, also should be followed carefully. Where a product is required to be protected from light and is in a clear or translucent container enclosed in an opaque outer covering, such outer covering is not to be removed and discarded until the contents have been used. In the absence of specific instructions, the product should be stored at controlled room temperature (see Storage Temperature in the General Notices). The product should be stored away from locations where excessive or variable heat, cold, or light prevails, such as those near heating pipes or fluorescent lighting.
Observing Products for Evidence of Instability—
Loss of potency usually results from a chemical change, the most common reactions being hydrolysis, oxidation-reduction, and photolysis. Chemical changes may also occur through interaction between ingredients within a product, or rarely between product and container. An apparent loss of potency in the active ingredient(s) may result from diffusion of the drug into, or its combination with, the surface of the container-closure system. An apparent gain in potency usually is caused by solvent evaporation or by leaching of materials from the container–closure system.
The chemical potency of the active ingredient(s) is required to remain within the limits specified in the monograph definition. Potency is determined by means of an assay procedure that differentiates between the intact molecule and its degradation products. Chemical stability data should be available from the manufacturer. Although chemical degradation ordinarily cannot be detected by the pharmacist, excessive chemical degradation sometimes is accompanied by observable physical changes. In addition, some physical changes not necessarily related to chemical potency, such as change in color and odor, formation of a precipitate, or clouding of solution, may serve to alert the pharmacist to the possibility of a stability problem. It should be assumed that a product that has undergone a physical change not explained in the labeling may also have undergone a chemical change, and such a product is never to be dispensed. Excessive microbial growth, contamination, or both, may also appear as a physical change. A gross change in a physical characteristic such as color or odor is a sign of instability in any product. Other common physical signs of deterioration of dosage forms include the following.
Solid Dosage Forms—
Many solid dosage forms are designed for storage under low-moisture conditions. They require protection from environmental water and therefore should be stored in tight containers (see Containers in the General Notices) or in the container supplied by the manufacturer. The appearance of fog or liquid droplets, or clumping of the product, inside the container signifies improper conditions. The presence of a desiccant inside the manufacturer's container indicates that special care should be taken in dispensing. Some degradation products, for example, salicylic acid from aspirin, may sublime and be deposited as crystals on the outside of the dosage form or on the walls of the container.
hard and soft gelatin capsules—
Since the capsule formulation is encased in a gelatin shell, a change in gross physical appearance or consistency, including hardening or softening of the shell, is the primary evidence of instability. Evidence of release of gas, such as a distended paper seal, is another sign of instability.
uncoated tablets—
Evidence of physical instability in uncoated tablets may be shown by excessive powder and/or pieces (i.e., crumbling as distinct from breakage) of tablet at the bottom of the container (from abraded, crushed, or broken tablets); cracks or chips in tablet surfaces; swelling; mottling; discoloration; fusion between tablets; or the appearance of crystals that obviously are not part of the tablet itself on the container walls or on the tablets.
coated tablets—
Evidence of physical instability in coated tablets is shown by cracks, mottling, or tackiness in the coating and the clumping of tablets.
dry powders and granules—
Dry powders and granules that are not intended for constitution into a liquid form in the original container may cake into hard masses or change color, which may render them unacceptable.
powders and granules intended for constitution as suspensions—
Dry powders and granules intended for constitution into solutions or suspensions require special attention. Usually such forms are antibiotics or vitamins that are particularly sensitive to moisture. Since they are always dispensed in the original container, they generally are not subject to contamination by moisture. However, an unusual caked appearance necessitates careful evaluation, and the presence of a fog or liquid droplets inside the container generally renders the preparation unfit for use. Presence of an objectionable odor also may be evidence of instability.
effervescent tablets, granules, and powders—
Effervescent products are particularly sensitive to moisture. Swelling of the mass or development of gas pressure is a specific sign of instability, indicating that some of the effervescent action has occurred prematurely.
Liquid Dosage Forms—
Of primary concern with respect to liquid dosage forms are homogeneity and freedom from excessive microbial contamination and growth. Instability may be indicated by cloudiness or precipitation in a solution, breaking of an emulsion, nonresuspendable caking of a suspension, or organoleptic changes. Microbial growth may be accompanied by discoloration, turbidity, or gas formation.
solutions, elixirs, and syrups—
Precipitation and evidence of microbial or chemical gas formation are the two major signs of instability.
emulsions—
The breaking of an emulsion (i.e., separation of an oil phase that is not easily dispersed) is a characteristic sign of instability; this is not to be confused with creaming, an easily redispersible separation of the oil phase that is a common occurrence with stable emulsions.
suspensions—
A caked solid phase that cannot be resuspended by a reasonable amount of shaking is a primary indication of instability in a suspension. The presence of relatively large particles may mean that excessive crystal growth has occurred.
tinctures and fluidextracts—
Tinctures, fluidextracts, and similar preparations usually are dark because they are concentrated, and thus they should be scrutinized carefully for evidence of precipitation.
sterile liquids—
Maintenance of sterility is of course critical for sterile liquids. The presence of microbial contamination in sterile liquids usually cannot be detected visually, but any haze, color change, cloudiness, surface film, particulate or flocculent matter, or gas formation is sufficient reason to suspect possible contamination. Clarity of sterile solutions intended for ophthalmic or parenteral use is of utmost importance. Evidence that the integrity of the seal has been violated on such products should make them suspect.
Semisolids (Creams, Ointments, and Suppositories)—
For creams, ointments, and suppositories, the primary indication of instability is often either discoloration or a noticeable change in consistency or odor.
creams—
Unlike ointments, creams usually are emulsions containing water and oil. Indications of instability in creams are emulsion breakage, crystal growth, shrinking due to evaporation of water, and gross microbial contamination.
ointments—
Common signs of instability in ointments are a change in consistency and excessive “bleeding” (i.e., separation of excessive amounts of liquid) and formation of granules or grittiness.
suppositories—
Excessive softening is the major indication of instability in suppositories, although some suppositories may dry out and harden or shrivel. Evidence of oil stains on packaging material should warn the pharmacist to examine individual suppositories more closely by removing any foil covering. As a general rule (although there are exceptions), suppositories should be stored in a refrigerator (see Storage Temperature in the General Notices).
Proper Treatment of Products Subjected to Additional Manipulations—
In repackaging, diluting a product or mixing it with another product, the pharmacist may become responsible for its stability.
Repackaging—
In general, repackaging is inadvisable. However, if repackaging is necessary, the manufacturer should be consulted concerning potential problems. In the filling of prescriptions, it is essential that suitable containers be used. Appropriate storage conditions and, when appropriate, an expiration date and beyond use date should be indicated on the label of the prescription container. Single-unit packaging calls for care and judgment and for strict observance of the following guidelines: (1) use appropriate packaging materials, (2) if stability data on the new package are not available, repackage at any one time only sufficient stock for a limited time, (3) include on the unit-dose label a lot number and an appropriate beyond-use date, (4) if a sterile product is repackaged from a multiple-dose vial into unit-dose (disposable) syringes, discard the latter if not used within 24 hours, unless data are available to support longer storage, (5) if quantities are repackaged in advance of immediate need, maintain suitable repackaging records showing name of manufacturer, lot number, date, and designation of persons responsible for repackaging and for checking (see General Notices), (6) if safety closures are required, use container closure systems that ensure compliance with compendial and regulatory standards for storage.
Dilution or Mixing—
If a product is diluted, or if two products are mixed, the pharmacist should observe good professional and scientific procedures to guard against incompatibility and instability. For example, tinctures such as those of belladonna and digitalis contain high concentrations of alcohol to dissolve the active ingredient(s), and they may develop a precipitate if they are diluted or mixed with aqueous systems. Pertinent technical literature and labeling should be consulted routinely; it should be current literature, because at times formulas are changed by the manufacturer. If a particular combination is commonly used, consultation with the manufacturer(s) is advisable. Since the chemical stability of extemporaneously prepared mixtures is unknown, the use of such combinations should be discouraged; if such a mixture involves an incompatibility, the pharmacist might be responsible. Oral antibiotic preparations constituted from powder into liquid form should never be mixed with other products.
Combining parenteral products necessitates special care, particularly in the case of intravenous solutions, primarily because of the route of administration. This area of practice demands the utmost in care, aseptic technique, judgment, and diligence. Because of potential unobservable problems with respect to sterility and chemical stability, all extemporaneous parenteral preparations should be used within 24 hours unless data are available to support longer storage.
Informing and Educating the Patient—
As a final step in meeting responsibility for the stability of drugs dispensed, the pharmacist is obligated to inform the patient about the proper storage conditions (for example, in a cool, dry place—not in the bathroom) for both prescription and nonprescription products, and to suggest a reasonable estimate of the time after which the medication should be discarded. When beyond-use dates are applied, the pharmacist should emphasize to the patient that the dates are applicable only when proper storage conditions are observed. Patients should be encouraged to clean out their drug storage cabinets periodically.
Auxiliary Information— Please check for your question in the FAQs before contacting USP.
| Topic/Question | Contact | Expert Committee |
| General Chapter | Desmond G. Hunt, Ph.D.
Scientist 1-301-816-8341 |
(PS05) Packaging and Storage 05 |
USP32–NF27 Page 706
Pharmacopeial Forum: Volume No. 28(1) Page 112