GENERAL GUIDANCES
Statistical Procedures for Bioequivalence Studies Using a Standard Two-Treatment Crossover Design2
introduction
The FDA Division of Bioequivalence in the Office of Generic Drugs usually evaluates bioequivalence by comparing the in vivo rate and extent of drug absorption of a test and reference formulation in healthy subjects. In a standard in vivo bioequivalence study design, study participants receive test and reference products on separate occasions, in either single or multiple doses, with random assignment to the two possible sequences of product administration. Samples of an accessible biologic fluid such as blood or urine are analyzed for drug and metabolite concentrations, and pharmacokinetic parameters (AUC, Cmax and Tmax) are obtained from the resulting concentration-time curves. These pharmacokinetic parameters are then analyzed statistically to determine if the test and reference products yield comparable values.
Standard statistical methodology based on the null hypothesis is not appropriate to assess bioequivalence. The Division of Bioequivalence has therefore employed a testing procedure termed the two one-sided tests procedure to determine whether average values for pharmacokinetic parameters measured after administration of the test and reference products are comparable. This procedure involves the calculation of a confidence interval for the ratio (or difference) between the test and reference product pharmacokinetic variable averages. The limits of the observed confidence interval must fall within a predetermined range for the ratio (or difference) of the product averages. The determination of the confidence interval range and the statistical level of significance are judgments made by the Division of Bioequivalence.
This guidance provides information about general pharmacokinetic and statistical analyses of bioequivalence data to be conducted by sponsors of abbreviated new drug and antibiotic applications and addresses three specific aspects of the statistical analysis as follows:
- Logarithmic transformation of pharmacokinetic data
- Sequence effect
- Outlier consideration.
This guidance became effective July 1, 1992. Any investigations initiated after this date should generally conform to the recommendations of the guidance. Sponsors following a different approach are encouraged to discuss the matter in advance with the FDA to prevent the expenditure of money and effort on preparing a submission that may later be determined to be unacceptable.
general methodology
Pharmacokinetic Analysis—
Single-Dose Studies—
At a minimum, the following pharmacokinetic parameters for the substances of interest should be measured in a single-dose bioequivalence study:
a. Area under the plasma/blood concentration-time curve from time zero to time t (AUC0–t), calculated by the trapezoidal rule, where t is the last measurable time point.
b. Area under the plasma/blood concentration-time curve from time zero to time infinity (AUC0– ), where AUC0– = AUCt + Ct /
), where AUC0– = AUCt + Ct /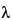 Z, Ct is the last measurable drug concentration, and Z is the terminal elimination rate constant calculated according to an appropriate method. The terminal or elimination half-life of the drug (t½) should also be reported.
Z, Ct is the last measurable drug concentration, and Z is the terminal elimination rate constant calculated according to an appropriate method. The terminal or elimination half-life of the drug (t½) should also be reported.
c. Peak drug concentration (Cmax) and the time to peak drug concentration (Tmax), obtained directly from the data without interpolation.
Multiple-Dose Studies—
At a minimum, the following pharmacokinetic parameters for the substances of interest should be measured in a multiple-dose bioequivalence study:
a. Area under the plasma/blood concentration-time curve from time zero to time 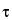 over a dosing interval at steady state (AUC0–), where is the dosing interval.
over a dosing interval at steady state (AUC0–), where is the dosing interval.
b. Peak drug concentration (Cmax) and the time to peak drug concentration (Tmax), obtained directly from the data without interpolation, after the last dose is administered.
c. Drug concentrations at the end of each dosing interval during steady state (Cmin).
d. Average drug concentration at steady state (Cav), where Cav = AUC0–/.
e. Degree of fluctuation (DF) at steady state, where DF = 100% × (Cmax – Cmin)/Cav.
Evidence of attainment of steady state for the test and reference products should be submitted in the bioequivalence study report.
Statistical Analysis—
Parametric (normal-theory) general linear model procedures are recommended for the analysis of pharmacokinetic data derived from in vivo bioequivalence studies. An analysis of variance (ANOVA) should be performed on the pharmacokinetic parameters AUC and Cmax using appropriate statistical programs and models. For example, for a conventional two-treatment, two-period, two-sequence (2 × 2) randomized crossover study design, the statistical model often includes factors accounting for the following sources of variation:
- Sequence (sometimes called Group or Order)
- Subjects, nested in sequences
- Period (or Phase)
- Treatment (sometimes called Drug or Formulation).
The sequence effect should be tested using the [subject (sequence)] mean square from the ANOVA as an error term. All other main effects should be tested against the residual error (error mean square) from the ANOVA. The least square means (LSMEANS) statement should be used to calculate least square means for treatments. Estimates should be obtained for the adjusted differences between treatment means and the standard error associated with these differences.
The two one-sided hypotheses at the 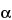 = 0.05 level of significance should be tested for AUC and Cmax by constructing the 90% confidence interval for the ratio between the test and reference averages.
= 0.05 level of significance should be tested for AUC and Cmax by constructing the 90% confidence interval for the ratio between the test and reference averages.
logarithmic transformation of pharmacokinetic data
Statistical Assumptions—
The assumptions underlying the ANOVA are:
- Randomization of samples
- Homogeneity of variances
- Additivity (linearity) of the statistical model
- Independency and normality of residuals.
In bioequivalence studies, these assumptions can be interpreted as follows:
- The subjects chosen for the study should be randomly assigned to the sequences of the study.
- The variances associated with the two treatments, as well as between the sequence groups, should be equal or at least comparable.
- The main effects of the statistical model, such as subject, sequence, period, and treatment effect for a standard 2 × 2 crossover study, should be additive. There should be no interactions between these effects.
- The residuals of the model should be independently and normally distributed. In other words, data from bioequivalence studies should have a normal distribution.
If these assumptions are not met, additional steps should be taken prior to the ANOVA including data transformation to improve the fit of the assumptions or use of a nonparametric statistical test in place of ANOVA. However, the normality and constant variance assumptions in the ANOVA model are known to be relatively robust [i.e., a small or moderate departure from each (or both) of these assumptions will not have a significant effect on the final result].
Rationale for Log Transformation—
It is acceptable to use logarithms to the base 10 or natural logarithms (ln). The report must state unambiguously which logarithms were used, and the use must be consistent throughout.
Clinical Rationale—
The primary comparison of interest in a bioequivalence study is the ratio of average parameter data from the test and reference formulations rather than the difference between them. Using log transformation, the general linear statistical model employed in the analysis of bioequivalence data allows inferences about the difference between the two means on the log scale, which can then be retransformed into inferences about the ratio of the two averages (means or medians) on the original scale. Log transformation thus achieves the general comparison based on the ratio rather than the difference.
Pharmacokinetic Rationale—
Westlake observed that a multiplicative model is postulated for pharmacokinetic parameters in bioavailability/bioequivalence studies, i.e., AUC and Cmax (but not Tmax). Assuming that elimination of the drug is first order and only occurs from the central compartment, the following equation holds after an extravascular route of administration:
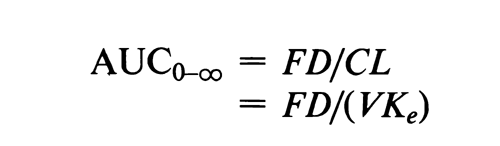 where F is the fraction absorbed, D is the administered dose, and FD is the amount of drug absorbed. CL is the clearance of a given subject, which is the product of the apparent volume of distribution (V) and the elimination rate constant (Ke).3
where F is the fraction absorbed, D is the administered dose, and FD is the amount of drug absorbed. CL is the clearance of a given subject, which is the product of the apparent volume of distribution (V) and the elimination rate constant (Ke).3
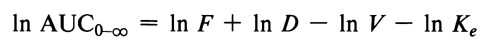
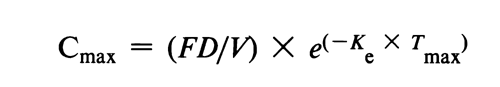

The use of AUC as a measure of the amount of drug absorbed thus involves a multiplicative term (CL), which might be regarded as a function of the subject. For this reason, Westlake contends that the subject effect is not additive if the data is analyzed on the original scale of measurement.
Logarithmic transformation of the AUC data will bring the CL(VKe) term into the equation in an additive fashion:
Similar arguments were given for Cmax. The following equation applies for a drug exhibiting one compartmental characteristic:
where again F, D, and V are introduced into the model in a multiplicative manner. However, after logarithmic transformation the equation becomes
Log transformation of the Cmax data also results in the additive treatment of the V term.
Statistical Rationale—
Logarithmic transformation of the data from bioequivalence studies can be used to circumvent the use of estimates of the reference product average for computation of the confidence interval for the ratio of product averages. This is an advantage for the cases where a least square estimate for the reference product mean is not well defined. Standard parametric methods are ill-suited to making inferences about the ratio of two averages, though some valid methods do exist. Log transformation changes the problem to one of making inferences about the difference (on the log scale) of two averages, for which the standard methods are well suited.
Many biological data correspond more closely to a log-normal distribution than to a normal distribution. The plasma concentration data, including the derived parameters AUC and Cmax, tend to be skewed, and their variances tend to increase with the means. Log transformation is likely to remedy this situation and make the variances independent of the mean. In addition, frequency distributions skewed to the left (with a long tail to the right) are often made more symmetrical by log transformation.
This argument is actually less persuasive than the argument based on the additivity of the statistical model because it is based largely on the between-subject distribution of AUC and Cmax values. For crossover studies, it is largely the within-subject distribution of values that determines the validity and efficiency of the standard parametric methods of analysis. Despite the arguments regarding the effect of log transformation on normality of bioequivalence data, it is recognized that the limited sample size (20–30 subjects) in a bioequivalence study precludes a reliable determination of the underlying normal distribution of the data set either with or without log transformation.
General Procedures—
Pharmacokinetic parameters AUC and Cmax should be log transformed. Firms are not encouraged to test for normality of data distribution after log transformation, nor should they employ normality of data distribution as a justification for carrying out the statistical analysis on the original scale. Robustness of a balanced study to non-normality of the data plus use of log transformation will be adequate in most cases.
If a firm believes that the data of a particular bioequivalence study should be statistically analyzed on the untransformed basis rather than the log scale, justification based upon a sound scientific rationale, as well as the statistical methods to be used, ought to be submitted to and reviewed by the FDA Division of Bioequivalence or a comparable regulatory authority.
Presentation of Data—
The drug concentration in biological fluid at each sampling time point should be furnished untransformed for all the subjects who participated in the study. The derived pharmacokinetic parameters should also be furnished untransformed. The mean, the standard deviation, and the coefficient of variation for each variable should be computed and tabulated in the final report.
To facilitate bioequivalence comparisons, pharmacokinetic parameters for each individual should be displayed in parallel for the formulations tested. In particular, for AUC and Cmax, the difference (T  R), ratio (T/R), and log of ratio (log T/R or ln T/R) between the test and reference values should be tabulated side by side for all the subjects. For each subject, the summary tables should indicate in which sequence (test, reference or reference, test) the subject received the product. Histograms showing the frequency distribution of the difference and ln ratio (or log ratio) for the major pharmacokinetic parameters (AUC and Cmax) are useful in the submission.
R), ratio (T/R), and log of ratio (log T/R or ln T/R) between the test and reference values should be tabulated side by side for all the subjects. For each subject, the summary tables should indicate in which sequence (test, reference or reference, test) the subject received the product. Histograms showing the frequency distribution of the difference and ln ratio (or log ratio) for the major pharmacokinetic parameters (AUC and Cmax) are useful in the submission.
In addition to the arithmetic mean for the test and reference products, the geometric means (antilog of the means of the logs), means of the logs, and standard deviations of the logs should be calculated for AUC and Cmax. All means, including arithmetic mean, geometric mean, and means of the logs, as well as standard deviations and coefficients of variation, are to be included in the report.
Equivalence Criteria—
For a broad range of drugs, the FDA Division of Bioequivalence used a range of 80% to 120% for the ratio of the product averages as the standard equivalence criterion when the study data are analyzed on the untransformed basis. This corresponds to a range of ±20% for the relative difference between the product averages.
When log-transformed data are used in the analysis of AUC and Cmax, using a range of 80% to 125% for the ratio of averages has an advantage over the 80% to 120% criterion in that for the analysis of log-transformed data, the probability of concluding equivalence is at a maximum if the ratio of averages is in fact 1.0 (i.e., exact equality). For the analysis of log-transformed data with a criterion of 80% to 120%, the maximum probability of concluding equivalence occurs when the ratio of product averages equals approximately 0.98. Thus, an equivalence criterion of 80% to 125% is used for the ratio of the product averages.
The 90% confidence interval for the difference in the means of the log-transformed data should be calculated using methods appropriate to the experimental design. The antilogs of the confidence limits constitute the 90% confidence interval for the ratio of the test and reference product averages.
sequence effect
A major limitation of a conventional two-treatment, two-period, two-sequence crossover design is the confounding between (i) a true sequence or group effect, (ii) unequal residual or carryover effects, and (iii) treatment-by-period interactions. A true sequence effect (i.e., a difference between the average response for sequence group one and sequence group two) would not bias the determination of bioequivalence. Unequal residual effects, however, would bias this estimate. A treatment-by-period interaction based on an underlying physical basis (i.e., if there were actually something about the periods that caused the difference between the product averages to differ from one period to another), would lead to difficulties in interpreting the estimate of the ratio (difference) in the pharmacokinetic parameters between the test and reference formulations.
Even if there were no true sequence effect, no unequal residual effects, and no treatment-by-period statistical interaction, approximately 10 out of every 100 two-treatment crossover studies would be likely to show an apparent sequence effect, if the testing is carried out at the 10% level of significance.
If the ANOVA test for the presence of a sequence effect results in statistical significance, the actual cause cannot be determined from the data alone. In some cases, plausible causes might be evaluated by examining demographic or physiological subject data, but this examination is seldom conclusive.
On the basis of these considerations, an in vivo two-treatment, two-period, two-sequence crossover bioequivalence study showing a statistically significant sequence effect may be acceptable provided:
- It is a single-dose study;
- It includes only healthy, normal subjects;
- The drug is not an endogenous entity;
- More than adequate washout period has been allowed between the two phases of the study, and in the second phase, the predose biological matrix samples do not exhibit any detectable drug level in all subjects; and
-
The study meets all scientific and statistical criteria such as:
- It is based upon an acceptable study protocol;
- It contains an acceptable validated assay methodology;
- The study data are acceptable;
- Appropriate statistical analyses of the data are performed; and
- Acceptable confidence intervals for the pharmacokinetic parameters are achieved.
Under all other circumstances, the sponsor may be asked to conduct another study. After regulatory review, multiple-dose studies or studies in patients demonstrating a statistically significant sequence effect may be acceptable provided they meet all other criteria listed above.
outlier consideration
Outliers are defined in bioequivalence studies as subjects having discordant values of one or more pharmacokinetic parameters when compared with other values in a study. Because bioequivalence studies are usually carried out as crossover studies, the most important type of outlier is where one or a few subjects differ notably from the rest of the subjects for the test product response versus the reference product response (e.g., test minus reference difference, test/reference ratio, or the log of the test/reference ratio).
The existence of an outlier could be indicative of the following problems with interchangeability of two formulations:
Product Failure—
A subject obtained an unusually high or low response to one or the other of the products because of a problem with the specific dosage unit(s) administered. Examples include a modified- or extended-release dosage form exhibiting dose dumping or a dosage unit whose coating inhibited dissolution.
Subpopulation—
A subject may be representative of a type of subject, present in the general population in low numbers, for whom the relative bioavailability of the two products is markedly different than it is for the majority of the population, and for whom the two products are not bioequivalent, even though they might be bioequivalent in the majority of the population.
In the case of product failure, it may make a difference whether the unusual response is observed on the test product or the reference product. In the case of a subpopulation, however, even if the unusual response is observed on the reference product, there may still be concern for lack of interchangeability of the two products.
Statistical tests exist for outlier identification. For detection of a single outlier, one important test is based on the absolute value of the “Studentized Residual.” Out of all the data in the study, the test focuses on the most extreme. Approximate critical values for this test have been published by Lund.9 In principle, however, outliers cannot be dropped from the analysis of the data solely on the basis of a statistical test. When one or more outliers are identified, one should provide scientific evidence or explanations to justify the exclusion of the subject data from statistical analysis.
Oral Extended-Release Dosage Forms4
This guidance describes in vivo bioequivalence studies and in vitro drug release testing recommended to applicants intending to submit Abbreviated New Drug Applications (ANDAs) for extended-release products administered orally.
nomenclature
Modified-Release Dosage Forms—
A modified-release dosage form is one for which the drug release characteristics of time course and/or location are chosen to accomplish therapeutic or convenience objectives not offered by conventional dosage forms such as solutions, ointments, or promptly dissolving dosage forms. Delayed-release and extended-release dosage forms are two types of modified-release dosage forms. This guidance does not consider bioequivalence studies for delayed-release formulations.
Delayed-Release Dosage Forms—
A delayed-release dosage form is one that releases a drug(s) at a time other than promptly after administration.
Extended-Release Dosage Forms—
An extended-release dosage form is one that allows at least a twofold reduction in dosing frequency or significant increase in patient compliance or therapeutic performance as compared to that presented as a conventional dosage form (e.g., as a solution or a prompt drug-releasing, conventional solid dosage form).
The terms controlled release, prolonged action, and sustained release are used synonymously with extended release. This document uses the term extended release to describe a formulation that does not release active drug substance immediately after oral dosing and that also allows a reduction in dosage frequency. This nomenclature accords generally with the USP definition of extended release but does not specify an impact on dosing frequency. The terms controlled release and extended release are considered interchangeable in this guidance.
regulatory background and general requirements
The Drug Price Competition and Patent Term Restoration Act amendments of 1984 to the Food, Drug, and Cosmetic Act gave the Food and Drug Administration statutory authority to accept and approve for marketing ANDAs for generic substitutes of innovator products, including those approved after 1962. To gain approval, ANDAs for a generic extended-release formulation must demonstrate, among other things, that the formulation is both pharmaceutically equivalent and bioequivalent to the innovator extended-release product, which is also termed the reference listed product as identified in FDA's Approved Drug Products with Therapeutic Equivalence Rating, “The Orange Book” (USP DI, Volume III).
Pharmaceutical Equivalence—
To be pharmaceutically equivalent, the generic and innovator formulations must (1) contain the same active ingredient; (2) contain the same strength of the active ingredient in the same dosage form; (3) be intended for the same route of administration, and (4) generally be labeled for the same conditions of use. The FDA does not require that the generic and reference listed extended-release products contain the same excipients or that the mechanism by which the active drug substance is released from the formulation be the same.
Bioequivalence Studies—
Current regulations require that bioequivalence be demonstrated between a generic extended-release formulation and the reference listed product. The reference listed product is generally an extended-release product subject to an approved full New Drug Application (NDA). For approval, documentation of bioequivalence must be established through performance of a series of in vivo bioequivalence studies that are defined under the section In Vivo Bioequivalence Studies for Approval. Approval of an ANDA will rely on data derived from evaluation of a biobatch, which is to be manufactured in accordance with the Office of Generic Drugs Procedure and Policy Guide 22-90.
Quality control of the manufacture of an extended-release formulation after approval may be assessed, in part, through performance of in vitro dissolution tests. Preapproval submission of these data is required. Recommendations for the conditions under which this test may be performed are described under the section In Vitro Dissolution for Quality Control Preapproval. This section also describes how specifications for this test are developed by the applicant and approved by the Division of Bioequivalence. These data are required in the application for approval.
in vivo bioequivalence studies for approval
In vivo bioequivalence studies recommended for approval for extended-release generic formulations are designed to document the following:
- The drug product meets the extended-release claim made for it.
- The drug product does not release the active drug substance at too rapid a rate (dose dumping).
- Performance is equivalent between the generic and the reference listed product following single doses and dosing to steady state.
- The influence of food on the vivo performance is comparable for the generic formulation relative to the innovator formulation.
The above objectives are generally met by three in vivo studies: (1) a single-dose, randomized, two-period, two-treatment, two-sequence crossover study under fasting conditions, comparing equal doses of the test and reference products; (2) a single-dose, randomized, three-treatment, three-period, six-sequence, crossover, limited food effects study, comparing equal doses of the test product administered under fasting conditions with those of the test and reference products administered immediately after a standard high-fat breakfast6; and (3) a multiple-dose, steady state, randomized, two-treatment, two-period, two-sequence crossover study under fasting conditions comparing equal doses of the test and reference formulations. [note—For safety reasons, this study may be performed in the nonfasting state. Applicants are encouraged to submit a study protocol describing the safety considerations requiring deviation from the fasting state to the Division of Bioequivalence for review prior to execution of the study.]
These studies are described in detail below. Under certain circumstances, the Division of Bioequivalence in the Office of Generic Drugs may require additional single-dose or multiple-dose steady state studies. The following general information relative to the three in vivo studies is provided:
- FDA-designated reference product is identified by the symbol “+” in The Orange Book.
- The assayed potency of the test product should not differ from that of the reference product by more than 5%.
- The clinical laboratory conducting any in vivo study should retain an appropriately identified reserve sample of the test and reference products for a period of 5 years. Each reserve sample should consist of at least 200 dosage units. For more information on retention of bioequivalence samples, refer to 21 CFR 320.63.
- A single-dose two-way crossover study under fasting conditions is required for each strength of a generic extended-release tablet formulation with multiple strengths. The multiple-dose steady state study and the fasting/nonfasting single-dose three-way crossover study are to be conducted with the highest strength only.
For an extended-release bead type capsule formulation to be marketed in multiple strengths, a single-dose bioequivalence study under fasting conditions is required only on the highest strength, provided that the compositions of the lower strengths are proportional to that of the highest strength, and the capsules contain identical beads or pellets. Single-dose in vivo bioequivalence studies may be waived for the lower strengths on the basis of acceptable drug release profiles. Multiple-dose steady state and single-dose fasting/nonfasting studies are to be conducted on the highest strength of the capsule formulation.
Batch Size—
The test batch or lot must be manufactured under production conditions and must be of a size that is at least 10% of the largest lot planned for full production or a minimum of 100,000 units, whichever is larger.
single-dose fasting two-way crossover bioequivalence study
Objective—
The objective is to compare the rate and extent of absorption of a generic extended-release product with that of the reference-listed product when administered in equal labeled doses.
Design—
The study design is a single-dose, two-treatment, two-period, two-sequence crossover with an adequate washout period (usually equal to at least 10 elimination half-lives of the drug) between the two phases of the study. An equal number of subjects should be randomly assigned to the two possible dosing sequences. Before the study begins, the proposed protocol should be approved by an institutional review board.
Facilities—
The clinical facilities and analytical laboratory used for the study should be identified along with the names, titles, and curriculum vitae of the medical and scientific or analytical directors.
Selection of Subjects—
The sponsor should enroll a number of subjects sufficient to ensure statistical validity of the study. It is recommended that a minimum of 24 subjects be used in this study. More subjects may be required for a drug that exhibits high intra-subject variability in metrics of rate and extent of absorption. Subjects should be healthy, preferably nonsmoking, volunteers 18 to 50 years of age, and within 10% of ideal body weight for height and build, although within 15% of ideal body weight is acceptable (Metropolitan Life Insurance Company Statistical Bulletin, 1983). Subjects should be accepted on the basis of acceptable medical history, physical examination, and clinical laboratory tests. Female subjects must be given a pregnancy test prior to beginning the study. Subjects with any current or past medical condition that might significantly affect their pharmacokinetic or pharmacodynamic response to the administered drug should be excluded from the study. If smokers are included, they should be identified as such. Written, informed consent must be obtained from all subjects before they are accepted into the study.
Procedure—
Following an overnight fast of at least 10 hours, subjects should be administered a single dose of the test or reference product with 240 mL of water. They should continue fasting for 4 hours after administration of the test or reference treatment.
Restrictions—
Study volunteers should observe the following restrictions:
- No alcohol or xanthine-containing foods or beverages should be consumed for 48 hours prior to dosing and until after the last blood sample is collected.
- Subjects should take no Rx medications, including oral contraceptives, beginning two weeks prior to and no OTC medications beginning one week prior to initiation of the study and until after the study is completed.
- Water may be taken except for 1 hour before and after administration, when no liquid is allowed other than that needed for drug dosing.
- All meals during the study should be standardized, and the same meals should be served during both phases of the study.
Blood Sampling—
In addition to the pre-dose (0 hour) sample, venous blood samples should be collected post-dose so that there are at least four sampling time points on the ascending part and six or more on the descending part of the concentration-time curve. The biological matrix (plasma, serum, or whole blood) should be immediately frozen after collection and, as appropriate, centrifugation, and kept frozen until assayed.
Subject Monitoring—
Blood pressure and pulse rate should be monitored hourly during the first 4 hours of the study. Subjects with a heart rate less than 45 bpm or greater than 110 bpm should have an electrocardiogram (lead II) performed and have their pulse monitored hourly. Subjects should report any unusual symptoms observed during the study. Subjects should be periodically questioned during each phase of the study for any unusual symptoms experienced after drug administration.
Analysis of Blood Samples—
The active ingredient should be assayed using a suitable analytical method validated with regard to specificity, accuracy, precision (both within and between days), limit of quantitation, linearity, and recovery. Stability of the samples under frozen conditions, at room temperature, and during freeze-thaw cycles, if appropriate, should be determined. If the analytical method is a chromatographic method, chromatograms of unknown samples, including all associated standard curve and quality control chromatograms, should be available for regulatory authorities.
Pharmacokinetic Analysis of Data—
Calculation of area under the plasma concentration-time curve to the last quantifiable concentration (AUC0–t) and to infinity (AUC0–), Cmax, and Tmax should be performed according to standard techniques.
Statistical Analysis of Pharmacokinetic Data (see Statistical Procedures for Bioequivalence Studies Using a Standard Two-treatment Crossover Design)—
The log transformed AUC and Cmax data should be analyzed statistically using ANOVA. These two parameters for the test product should be shown to be within 80% to 125% of the reference product using the 90% confidence interval.
Clinical Report, Side Effects, and Adverse Reactions—
Subject medical histories, physical examination and laboratory reports, and all incidents of possible adverse reactions to the study formulations should be reported.
multiple-dose steady state, two-way crossover bioequivalence study under fasting conditions
Objective—
The objective is to compare the steady-state rate and extent of absorption of a generic extended-release formulation with that of the reference formulation when given as equal labeled doses.
Design—
The study design is a multiple-dose, steady-state two-treatment, two-period, two-sequence crossover with an adequate washout period between the two phases of the study. An equal number of subjects should be randomly assigned to the two possible dosing sequences. Before beginning the study, the study protocol should be approved by an institutional review board.
Facilities and Selection of Subjects—
See the appropriate section under Single-Dose Fasting Two-Way Crossover Bioequivalence Study.
Procedure—
Extended-release products that are administered once a day should be dosed following an overnight fast of at least 10 hours; subjects should continue fasting for 4 hours post-dose. For extended-release products that are dosed every 12 hours (b.i.d.), the morning dose should be given following an overnight fast of about 10 hours, and subjects should continue fasting for 4 hours post-dose; the evening dose should be administered 12 hours after the morning dose and after a fast of at least 2 hours and subjects should continue fasting for 2 hours post-dose. Each dose should be administered with 240 mL of water.
Restrictions—
Study volunteers should observe the following restrictions:
- No alcohol or xanthine-containing foods or beverages should be consumed for 48 hours prior to dosing and until after the last blood sample is collected.
- Subjects should take no Rx medications, including oral contraceptives, beginning two weeks prior to and no OTC medications beginning one week prior to initiation of the study and until after the study is completed.
- Water may be taken except for 1 hour before and after administration, when no liquid is allowed other than that needed for drug dosing.
Blood Sampling—
At least three trough concentrations (Cmin) on three consecutive days should be determined to ascertain that the subjects are at steady state prior to measurement of rate and extent of absorption after a single-dose administration in a dosing interval at steady state. The three consecutive trough samples should be collected at the same time of the day and should be comparable. For extended-release drug products administered more often than every 24 hours, assessment of trough levels just prior to 2 consecutive doses is not recommended because a difference in the consecutive trough values may occur due to circadian rhythm irrespective of whether or not steady state has been attained. Adequate blood samples should be collected at appropriate times during a dosing interval at steady state to permit estimation of the total area under the concentration-time curve, peak concentration (Cmax), and time to peak concentration (Tmax).
Subject Monitoring and Analysis of Blood Samples—
See under Single-Dose Fasting Two-Way Crossover Bioequivalence Study.
Pharmacokinetic Analysis of Data—
The following pharmacokinetic data are to be reported for the evaluation of bioequivalence of the generic extended-release product with the reference listed product:
- Individual and mean blood drug concentration levels
- Individual and mean trough levels (Cmin)
- Individual and mean peak levels (Cmax)
- Calculation of individual and mean steady state AUCinterdose are recommended (AUCinterdose is AUC during a dosing interval at steady state)
- Individual and mean percent fluctuation [= 100 × (Cmax – Cmin)/Cmin]
- Individual and mean time to peak concentration (Tmax)
Statistical Analysis of Pharmacokinetic Data—
The log transformed AUC and Cmax data should be analyzed statistically using ANOVA. These two parameters for the test product should be shown to be within 80% to 125% of the reference product using the 90% confidence interval. Fluctuation for the test product should be evaluated for comparability with that for the reference product. For further information on statistical analysis, see Statistical Procedures for Bioequivalence Studies Using a Standard Two-Treatment Crossover Design.
Clinical Report, Side Effects, and Adverse Reactions—
See under Single-Dose Fasting Two-Way Crossover Bioequivalence Study.
single-dose, three-way crossover fasting/nonfasting bioequivalence study
Objective—
The objective is to compare the rate and extent of absorption of a generic formulation with that of the reference listed formulation under nonfasting conditions and to compare the rate and extent of absorption of the drug from a generic formulation under fasting and nonfasting conditions when given as equal labeled doses.
Design—
The study design is a single-dose, three-treatment, three-period, six-sequence crossover with adequate washout period between the three phases of the study. An equal number of subjects should be randomly assigned to each dosing sequence. Before beginning the study, the study protocol should be approved by an institutional review board.
Facilities—
See this section under Single-Dose Fasting Two-Way Crossover Bioequivalence Study.
Selection of Subjects—
A minimum of 18 subjects should be enrolled in this study. For other information on selection of subjects, see Single-Dose Fasting Two-Way Crossover Bioequivalence Study.
Procedure—
Each subject should receive the following three treatments:
treatment 1: Generic extended-release product administered after a high fat content breakfast
treatment 2: Innovator extended-release product (reference listed drug) administered after a high fat content breakfast.
treatment 3: Generic extended-release product administered after fasting.
treatment 1: Generic extended-release product administered after a high fat content breakfast
treatment 2: Innovator extended-release product (reference listed drug) administered after a high fat content breakfast.
treatment 3: Generic extended-release product administered after fasting.
Following an overnight fast of at least 10 hours, subjects receiving treatments under nonfasting conditions should be served a high-fat breakfast then immediately dosed with Treatment 1 or Treatment 2 with 240 mL of water. Subjects receiving Treatment 3 should be dosed at the same time as Treatments 1 and 2 with 240 mL of water only. No food should be allowed for at least 4 hours post-dose, with water allowed after the first hour. Subjects should be served scheduled standardized meals throughout the study, and the same meals should be served during all phases of the study.
Restrictions, Blood Sampling, Subject Monitoring, and Analysis of Blood Samples—
See the appropriate section under Single-Dose Fasting Two-Way Crossover Bioequivalence Study.
Statistical Analysis of Pharmacokinetic Data—
In general a comparable food effect will be assumed if the mean values of AUC0–t, AUC0–, and Cmax for the generic product administered with food differ by no more than 20% from the respective mean values for the reference listed product administered with food in the study.
Clinical Report, Side Effects, and Adverse Reactions—
See under Single-dose Fasting Two-way Crossover Bioequivalence Study.
in vitro drug release for quality control preapproval
Drug Release Testing  724
724 —
Drug release testing should be conducted on 12 individual dosage units of the batches of the test and reference products used in the bioequivalence studies. The potential for pH dependence of drug release from an extended-release product is well recognized. Drug release profiles should therefore be generated in aqueous media at the following pH ranges: 1-1.5, 4-4.5, and 6.0-6.8. Early sampling times of 1, 2, and 4 hours should be included in the sampling schedule to provide assurance against premature release of the drug (dose dumping) from the formulation. Typical drug release conditions are shown below:
—
Drug release testing should be conducted on 12 individual dosage units of the batches of the test and reference products used in the bioequivalence studies. The potential for pH dependence of drug release from an extended-release product is well recognized. Drug release profiles should therefore be generated in aqueous media at the following pH ranges: 1-1.5, 4-4.5, and 6.0-6.8. Early sampling times of 1, 2, and 4 hours should be included in the sampling schedule to provide assurance against premature release of the drug (dose dumping) from the formulation. Typical drug release conditions are shown below:
Apparatus 1 (for capsules):
100 rpm.
Apparatus 2 (for tablets):
50 and 75 rpm.
Temperature:
37 ± 0.5 .
.
Units Tested:
12.
Medium:
900 mL of aqueous media at various pH values.
Times:
1, 2, and 4 hours, and every 2 hours thereafter, until 80% of the drug is released.
Tolerances:
to be established based on data generated.
Content Uniformity Test 905—
Content uniformity testing on the test and reference product lots should be performed as described in USP.
Specifications—
Specifications for the drug release procedure to ensure quality control will be determined on a case-by-case basis. In general, further validation will be required to expand dissolution specifications beyond those established for the biobatch.