Add the following:
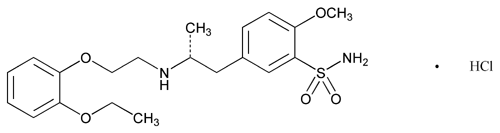
C20H28N2O5S·HCl
 444.97
444.97
Benzenesulfonamide, 5-[2-[[2-(2-ethoxyphenoxy)ethyl]amino]propyl]-2-methoxy-, monohydrochloride, (R)-.
(-)-(R)-5-[2-[[2-(o-Ethoxyphenoxy)ethyl]amino]propyl]-2-methoxybenzenesulfonamide monohydrochloride.
» Tamsulosin Hydrochloride contains not less than 98.5 percent and not more than 101.0 percent of C20H28N2O5S·HCl, calculated on the dried basis.
Packaging and storage—
Preserve in tight containers. Store at controlled room temperature.
USP Reference standards  11
11 —
—
USP Tamsulosin Hydrochloride RS.
USP Racemic Tamsulosin Hydrochloride RS.
USP Tamsulosin Hydrochloride RS.
USP Racemic Tamsulosin Hydrochloride RS.
Identification—
B:
Prepare a solution in water containing about 7.5 mg per mL. [Note—Use heating to dissolve the sample.] Cool in an ice bath 5 mL of the solution, add 3 mL of diluted nitric acid, and shake until mixed thoroughly. Allow the mixture to stand for 30 minutes at room temperature, and filter. The filtrate responds to the tests for Chloride 191.
Specific rotation 781S:
between  17.5
17.5 and 20.5, measured at 20.
and 20.5, measured at 20.
Test solution—
To 150 mg of sample, previously dried at 105 for 2 hours, add 20 mL of water, dissolve by heating at 60 to 70, and allow to cool.
Loss on drying 731—
Dry it at 105 for 2 hours: it loses not more than 0.5% of its weight.
Residue on ignition 281:
not more than 0.10%.
Heavy metals, Method II 231:
not more than 20 ppm.
Related substances—
Test 1: Impurities eluting before tamsulosin—
Solution pH 2.0—
Dissolve 8.7 mL of perchloric acid (70%) and 3.0 g of sodium hydroxide in 1900 mL of water. Adjust to a pH of 2.0 with 1 N sodium hydroxide, and add sufficient water to make 2000 mL.
Mobile phase 1—
Prepare a mixture of Solution pH 2.0 and acetonitrile (7:3). Make adjustments if necessary (see System Suitability under Chromatography 621).
Diluent—
Use Mobile phase 1.
Test solution—
Dissolve an accurately weighed portion of Tamsulosin Hydrochloride in Diluent to obtain a solution having a known concentration of about 5.0 mg per mL.
Standard solution—
Dilute a portion of the Test solution with Diluent to obtain a solution having a known concentration of 10 µg per mL.
System suitability solution—
Dissolve 5 mg of Tamsulosin Hydrochloride and 0.01 g of propylparaben in 20 mL of Diluent. Dilute 2 mL of this solution with Diluent to make 20 mL.
Chromatographic system (see Chromatography 621—
The liquid chromatograph is equipped with a 225-nm UV detector and a 4.6-mm × 15-cm column that contains 5-µm packing L1. The column temperature is maintained at 40. The flow rate is about 1.3 mL per minute. Chromatograph the System suitability solution, and record the peak responses as directed for Procedure: the elution order is tamsulosin hydrochloride followed by propylparaben, and the resolution between tamsulosin and propylparaben is not less than 12. Chromatograph the Standard solution: the relative standard deviation for six replicate injections is not more than 4%.
Procedure—
Inject equal volumes (about 10 µL) of the Standard solution and the Test solution into the chromatograph, record the chromatograms for about 1.5 times the retention time of tamsulosin, and measure the peak responses. Calculate the percentage of any individual impurity in the portion of Tamsulosin Hydrochloride taken by the formula:
100 (CS / CT)(rU / rS)
where CS and CT are the concentrations, in mg per mL, of tamsulosin in the Standard solution and the Test solution, respectively; rU is the area of each peak eluted before tamsulosin obtained from the Test solution; and rS is the peak area of tamsulosin obtained from the Standard solution. Not more than 0.10% of each impurity is found. [Note—If present, the des-ethoxy impurity and the methoxy impurity eluting at the relative retention time of about 0.8 are not separated by this method and should be integrated together to determine conformance. (The des-ethoxy impurity is 2-methoxy-5-[(2R)-2-[(2-phenoxyethyl)amino]propyl]benzenesulfonamide, and the methoxy impurity is 2-methoxy-5-[(2R)-2-[[2-(2-methoxyphenoxy)ethyl]amino]propyl]benzenesulfonamide.) Not more than 0.20% of the sum of the des-ethoxy and methoxy impurities is found.] The reporting level for impurities is 0.05%.
Test 2: Impurities eluting after tamsulosin—
Solution pH 2.0, Diluent, Test solution, and Standard solution—
Prepare as directed in Test 1 for Related compounds.
Mobile phase 2—
Prepare a mixture of Solution pH 2.0 and acetonitrile (1:1). Make adjustments if necessary (see System Suitability under Chromatography 621).
Chromatographic system (see Chromatography 621—
The liquid chromatograph is equipped with a 225-nm detector and a 4.6-mm × 15-cm column that contains 5-µm packing L1. The column temperature is maintained at 40. The flow rate is about 1.0 mL per minute. The relative standard deviation of six replicate injections of the Standard solution is not more than 4%. Use a column that meets the resolution requirements of Test 1 for Related compounds.
Procedure—
Inject a volume (about 10 µL) of the Standard solution and the Test solution into the chromatograph, record the chromatograms for at least 5 times the retention time of tamsulosin, and measure the peak responses. Calculate the percentage of any individual impurity in the portion of Tamsulosin Hydrochloride taken by the formula:
100 (CS / CT)(rU / rS)
where CS and CT are the concentrations, in mg per mL, of tamsulosin in the Standard solution and the Test solution, respectively; rU is the area of each peak eluted after tamsulosin obtained from the Test solution; and rS is the peak area of tamsulosin obtained from the Standard solution. Not more than 0.10% of each impurity is found. The reporting level for impurities is 0.05%. Not more than 0.2% of total impurities is found, the results for Test 1 and Test 2 being combined.
Enantiomeric purity—
Mobile phase—
Prepare a mixture of hexane, dehydrated alcohol, methanol, and diethylamine (650:200:150:1). Make adjustments if necessary (see System Suitability under Chromatography 621).
Test solution—
Dissolve an accurately weighed portion of Tamsulosin Hydrochloride in methanol to obtain a solution having a known concentration of about 2.0 mg per mL.
Standard solution—
Dilute a portion of the Test solution with methanol to obtain a solution having a known concentration of about 0.002 mg per mL.
System suitability solution—
Dissolve an accurately weighed portion of USP Racemic Tamsulosin Hydrochloride RS in methanol to obtain a solution having a known concentration of about 0.04 mg per mL.
Chromatographic system (see Chromatography 621)—
The liquid chromatograph is equipped with a 225-nm detector and a 4.6-mm × 25-cm column that contains packing L51. The column temperature is maintained at 40. The flow rate is about 0.5 mL per minute. Chromatograph the System suitability solution, and identify the components of the mixture on the basis of their relative retention times, which are about 0.8 for the optical isomer and 1.0 for tamsulosin. Record the peak responses as directed for Procedure: the resolution between the optical isomer and tamsulosin is not less than 2.
Procedure—
Separately inject equal volumes (about 10 µL) of the Standard solution and the Test solution into the chromatograph, record the chromatograms, and measure the peak responses. Calculate the percentage of the optical isomer in the portion of Tamsulosin Hydrochloride taken by the formula:
100 (CS / CT)(rU / rS)
where CS and CT are the concentrations, in mg per mL, of tamsulosin in the Standard solution and the Test solution, respectively; rU is the area of the optical isomer obtained from the Test solution; and rS is the peak area of tamsulosin obtained from the Standard solution. Not more than 0.3% of the optical isomer is found.
Assay—
Accurately weigh about 350 mg of Tamsulosin Hydrochloride, previously dried at 105 for 2 hours, and add 5 mL of formic acid to dissolve. Add 75 mL of a mixture of glacial acetic acid and acetic anhydride (3:2) to the resultant solution, and titrate immediately with 0.1 N perchloric acid VS, determining the endpoint potentiometrically. Perform a blank determination, and make any necessary corrections. Each mL of 0.1 N perchloric acid is equivalent to 44.50 mg of C20H28N2O5S·HCl. USP32
USP32
Auxiliary Information—
Please check for your question in the FAQs before contacting USP.
Chromatographic Column—
| Topic/Question | Contact | Expert Committee |
| Monograph | Elena Gonikberg, Ph.D.
Senior Scientist 1-301-816-8251 |
(MDGRE05) Monograph Development-Gastrointestinal Renal and Endocrine |
| Reference Standards | Lili Wang, Technical Services Scientist 1-301-816-8129 RSTech@usp.org |
USP32–NF27 Page 3652
Pharmacopeial Forum: Volume No. 33(6) Page 1211
Chromatographic columns text is not derived from, and not part of, USP 32 or NF 27.